Cuaderno de Ciencias
Miércoles 20 de octubre 10:54
Por Miguel Artime
La vía láctea actúa como si tuviera todo el tiempo del mundo por delante. Sus brazos en espiral giran alrededor de su punto central, y a pesar de que en algunos de estos brazos nacen nuevas estrellas a partir del gas que cae como lluvia fina desde el espacio intergaláctico, lo cierto es que la galaxia - y el universo - también encontrarán su final.
Nuestro sol y otras estrellas similares morirán un buen día, mucho antes que la galaxia, pero de momento en las calderas estelares de Orión y Taurus siguen naciendo nuevas estrellas que las sustituyen.
Otras estrellas más grandes desaparecerán como supernovas, pero la mayoría de las estrellas perecerán modestamente dejando atrás brasas que palidecerán con el tiempo. De hecho, la vida de una estrella viene determinada por su masa. Por norma general, las pequeñas viven más tiempo.
Pero ¿cómo será el final de nuestra galaxia?
Se especula que llegará despacio, a medida que la masa que compone las estrellas quede encarcelada. Las estrellas grandes viven deprisa y mueren de forma explosiva creando supernovas. Tras ellas queda una estrella de neutrones o un agujero negro, ambos cuerpos sin capacidad de emitir luz.
Las estrellas similares al Sol y las que tienen menos masa, morirán dejando tras de sí enanas blancas, que vienen a ser brasas ricas en carbono que van enfriándose lentamente. Gradualmente el ciclo de vida y muerte de las estrellas se romperá de forma irrevocable. Con el tiempo más y más masa quedará atrapada en restos estelares compactos, o en enanas blancas que se enfrían.
En el resto de las galaxias el escenario será similar. Poco a poco la luz del universo irá apagándose gradualmente. Después de miles de billones de años todo el universo habrá sufrido un fundido en negro. Y sin embargo, por extraño que parezca, el fin de la luz no implicará el fin de la vida.
Las estrellas brillan convirtiendo en radiación una pequeña porción de la energía encerrada en la materia que las compone. Cuando su luz se apague, la fuente final de luz estelar será la energía gravitatoria.
Y es que aparte de la fusión nuclear, existen otras formas de convertir la energía gravitatoria en calor o radiación, por lo que incluso después de que todas las estrellas hayan palidecido, las civilizaciones que aún pululen por el universo en esos fatídicos tiempos, podrán sobrevivir recolectando la energía de los agujeros negros.
Aprovechando esa energía y si la nostalgia de una estrella la llevase a ese punto, esa anciana civilización podría crear estrellas artificiales.
Y todavía no hemos hablado de la energía oscura, la cual según los físicos es la culpable de que la expansión del cosmos se esté acelerando. La energía oscura es una manifestación del vacío puro del espacio que provoca un efecto opuesto a la gravedad: repele en vez de atraer.
Para las teorías fundamentales la existencia de la energía oscura supuso algo embarazoso ya que ninguna la había predicho y hasta el momento nadie sabe cómo puede el vacío puro contar con una propiedad tan extraña.
Hay quien piensa que la energía oscura varía con el tiempo y el espacio. Si la energía oscura crece, hará que el tejido del universo se desenmarañe en aproximadamente 20 billones de años en un punto llamado "gran desgarramiento".
Si esta teoría es cierta, esto haría que primero las galaxias, luego las estrellas y al final los propios átomos, se rompiesen por la acción de la energía oscura. Como resultado de esta fatalidad, nada podría sobrevivir.
En ausencia del gran desgarramiento, la aceleración cósmica irá retirando a las galaxias de nuestra vista a un ritmo regular. Después de 100 billones de años, la mayoría de las galaxias se retirarán a una velocidad superior a la de la velocidad de la luz, dejando tras ellas imágenes congeladas finales del borde de nuestro horizonte.
Las Vías Láctea y Andrómeda se mezclarán y nuestra visión del universo acabará en el borde de esta supergalaxia.
A escalas de tiempo aún más largas, las familiares estructuras gravitatorias que nos enseñan en el colegio terminarán por despegarse. Cuando pasen de 10^15 años, los planetas acabarán por separarse de sus estrellas muertas y navegarán a la deriva por el espacio interestelar.
En 10^19 años las estrellas se separarán de sus galaxias y flotarán por el espacio intergaláctico.
Finalmente según la mayoría de las teorías que unifican las partículas fundamentales en términos de una única súper-fuerza, pasados 10^35 años el protón dejará de ser estable y terminará por descomponerse.
Para que nos demos cuenta de lo vasta de esta última escala de tiempo, podemos decir que en esos términos el tiempo transcurrido desde el origen del universo hasta la actualidad representa sólo un milisegundo.
La descomposición de los protones será el heraldo que anunciará una larga fase final de desintegración del universo en el que todo se viene abajo. Tras la descomposición de los protones, el átomo estable pasará a mejor vida, lo cual supone un reto mayúsculo para las formas de vida (si es que aún siguen ahí).
Finalmente caerá el telón con la lenta evaporación de los agujeros negros mediante un proceso llamado radiación de Hawking. Los más grandes terminarán por evaporarse transcurrida una inconcebible escala de tiempo de 10^98 años.
Podemos imaginar los últimos habitantes de ese ancianísimo universo a punto de expirar, arremolinados alrededor del fulgor de los rayos gamma que se evaporan del último agujero negro, contando historias infinitas acerca de lo que un día fue un universo que bullía en luz y vida.
Fue bonito mientras duró.
martes, 14 de diciembre de 2010
EL DESCUBRIMIENTO DE UN "BICHO DE ARSÉNICO" EXPANDE LA DEFINICIÓN DE LA VIDA

Diciembre 2, 2010: Con el apoyo de la NASA, un grupo de investigadores ha descubierto el primer microorganismo terrestre capaz de desarrollarse y reproducirse utilizando arsénico, un elemento químico muy tóxico. El microorganismo, que vive en el Lago Mono, California, sustituye al fósforo por arsénico para construir su ADN y otros componentes celulares.
"La definición de la vida acaba de expandirse", dijo Ed Weiler, administrador asociado de la NASA para el Directorio de Misiones Científicas, en las oficinas centrales de la agencia, ubicadas en Washington. "Conforme avanzamos en nuestros esfuerzos por encontrar signos de vida en el sistema solar, tenemos que ampliar nuestro pensamiento, hacerlo más diverso y considerar que puede existir vida de una manera diferente a la que conocemos".
El hallazgo de una composición bioquímica alternativa alterará los libros de texto de biología y expandirá el alcance de la búsqueda de vida fuera del planeta Tierra. La investigación será publicada en la edición de esta semana de la revista Science Express.
Carbono, hidrógeno, nitrógeno, oxígeno, fósforo y azufre son las seis piezas básicas de todas las formas de vida conocidas en la Tierra. El fósforo es parte de la columna vertebral química del ADN y del ARN, las estructuras que transportan las instrucciones genéticas para la vida, y es considerado un elemento esencial para todas las células vivas.
El fósforo es un componente esencial de la molécula que transporta la energía en todas las células (el adenosín trifosfato) y también de los fosfolípidos que conforman todas las membranas celulares. El arsénico, aunque es químicamente similar al fósforo, es venenoso para la mayoría de los seres vivos en la Tierra. El arsénico destruye los senderos del metabolismo porque, químicamente, se comporta de manera similar al fosfato.
"Sabemos que algunos microbios pueden respirar arsénico, pero lo que encontramos es un microbio que hace algo completamente distinto: construye partes de sí mismo con el arsénico", dijo Felisa Wolfe-Simon, una becaria de Investigación en Astrobiología para la NASA, en el Centro de Estudios Geológicos de Estados Unidos (U.S. Geological Survey, en idioma inglés), en Menlo Park, California, y quien dirigió al equipo que llevó a cabo la investigación. "Si algo aquí en la Tierra puede hacer algo tan inesperado, ¿qué más puede hacer la vida que aún no hemos visto?"
El microbio recién descubierto, la cepa GFAJ-1, es miembro de un grupo común de bacterias, las Gammaproteobacterias. En el laboratorio, los investigadores lograron exitosamente que los microbios del lago crecieran con una dieta muy baja en fósforo y muy generosa en arsénico. Cuando los investigadores quitaron el fósforo y lo reemplazaron con arsénico, los microbios continuaron creciendo. Análisis posteriores indicaron que el arsénico estaba siendo usado para producir los componentes básicos de las nuevas células de GFAJ-1.
El punto crucial que los científicos investigaron consistió en saber, para el microbio que creció en el arsénico, cuándo fue el arsénico incorporado en la maquinaria bioquímica vital del organismo, como lo es el ADN, las proteínas y las membranas celulares. Una variedad de técnicas sofisticadas de laboratorio fueron usadas para determinar dónde fue incorporado el arsénico.
El equipo escogió explorar el Lago Mono debido a su química inusual, especialmente su alta salinidad, su alta alcalinidad y sus altos niveles de arsénico. Esta química es, en parte, una consecuencia del aislamiento del Lago Mono de sus fuentes de agua dulce durante un período de 50 años.
Los resultados de este estudio aportarán información valiosa a investigaciones en muchas áreas, incluyendo el estudio de la evolución de la Tierra, la química orgánica, los ciclos bioquímicos, la lucha contra las enfermedades y la investigación de la Tierra como sistema. Estos hallazgos también abren nuevas fronteras en el campo de la microbiología y de otras áreas del conocimiento.
"La idea de bioquímicas alternativas para la vida es común en la ciencia ficción", dijo Carl Pilcher, quien es el director del Instituto de Astrobiología de la NASA (NASA Astrobiology Institute, en idioma inglés), en el Centro de Investigaciones Ames (Ames Research Center, en idioma inglés), ubicado en Moffet Field, California. "Hasta ahora, una forma de vida que usa el arsénico como bloque fundamental era solamente teoría, pero ahora sabemos que un tipo de vida como ese existe en el Lago Mono".
El equipo de investigación incluye a científicos del Centro de Estudios Geológicos de Estados Unidos, de la Universidad Estatal de Arizona (Arizona State University, en idioma inglés), localizado en Tempe, Arizona, del Laboratorio Nacional Lawrence Livermore (Lawrence Livermore National Laboratory, en idioma inglés), ubicado en Livermore, California, de la Universidad Duquesne (Duquesne University, en idioma inglés), en Pittsburgh, Pennsilvannia, y de la Fuente de Radiación Sincrotrónica de Stanford (Standford Synchroton Radiation LightSource, en idioma inglés), en Menlo Park, California.
El Programa de Astrobiología de la NASA (NASA Astrobiology Program, en idioma inglés), en Washington, contribuyó con fondos para las investigaciones a través de su Programa de Exobiología y Biología Evolutiva (Exobiology and Evolutionary Biology, en idioma inglés), así como del Instituto de Astrobiología (Institute for Astrobiology, en idioma inglés), de la NASA. El Programa de Astrobiología de la NASA apoya investigaciones sobre el origen, la evolución, la distribución y el futuro de la vida en la Tierra.
Créditos y Contactos
Autor: Dr. Tony Phillips
Funcionaria Responsable de NASA: Ruth Netting
Editor de Producción: Dr. Tony Phillips Traducción al Español: Carlos Román
Editora en Español: Angela Atadía de Borghetti
Formato: Carlos Román Zúñiga
lunes, 25 de octubre de 2010
Mecanismos de Acción de los Antimicrobianos

Por Jorge Calvo a, Luis Martínez-Martínez a
Servicio de Microbiología, Hospital Universitario Marqués de Valdecilla, Santander, España.
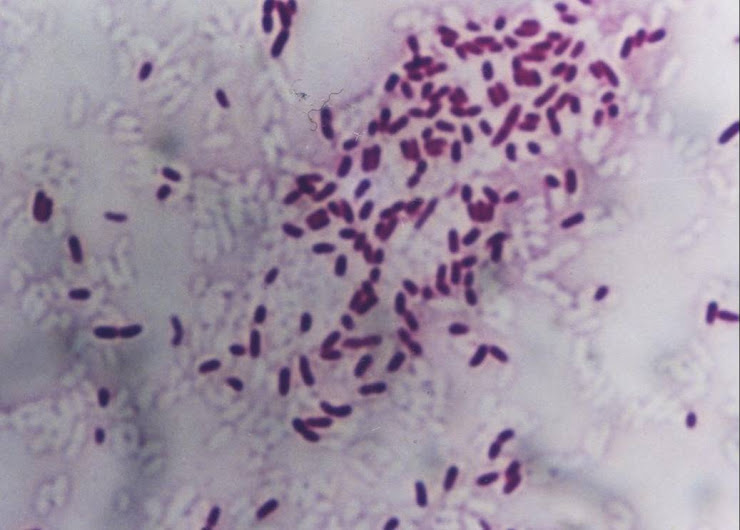
Hay una amplia diversidad de familias y grupos de antimicrobianos de interés clínico. Los mecanismos por los que los compuestos con actividad antibacteriana inhiben el crecimiento o causan la muerte de las bacterias son muy variados, y dependen de las dianas afectadas. La pared celular (una estructura singular de la inmensa mayoría de las bacterias, ausente en células eucariotas) puede verse afectada en la síntesis (fosfomicina, cicloserina) o el transporte de sus precursores (bacitracina, mureidomicinas), o en su organización estructural (β-lactámicos, glucopéptidos). Los principales derivados que afectan a la membrana citoplásmica son las polimixinas y la daptomicina. La síntesis proteica puede bloquearse por una amplia variedad estructural de compuestos que afectan a algunas de las fases de este proceso: activación (mupirocina), iniciación (oxazolidinonas, aminoglucósidos), fijación del complejo aminoácido-ARNt al ribosoma (tetraciclinas, glicilciclinas) o elongación (anfenicoles, lincosamidas, macrólidos, cetólidos, estreptograminas o ácido fusídico). El metabolismo de los ácidos nucleicos puede verse afectado en la ARN polimerasa dependiente de ADN (rifamicinas) o en el proceso de enrollamiento/desenrollamiento del ADN (quinolonas); algunos compuestos afectan directamente al ADN (nitroimidazoles, nitrofuranos). El trimetoprim y las sulfamidas (con frecuencia usados en combinación) son los representantes de los antimicrobianos que bloquean las vías metabólicas de la bacteria. Algunos compuestos, aun siendo incapaces de inhibir o matar las bacterias, pueden bloquear sus mecanismos de resistencia, por lo que usados en combinación con otros antimicrobianos potencian la acción de estos últimos; de este grupo de sustancias sólo se emplean en clínica algunos inhibidores de β-lactamasas.
Enferm Infecc Microbiol Clin. 2009;27:44-52.
Palabras clave: Antimicrobianos. Mecanismo de acción. Pared cellular. Membrana citoplásmica. Síntesis proteica. ADN. Vías metabólicas.
IntroducciónEl descubrimiento de la penicilina en 19291 y su posterior introducción en clínica supuso una verdadera revolución en el tratamiento de la patología infecciosa. Desde entonces, se han incorporado a la práctica clínica decenas de familias de antimicrobianos, con actividad frente a bacterias, hongos, parásitos y virus. En este artículo se abordarán los aspectos microbiológicos del mecanismo de acción de los compuestos con actividad antibacteriana.
Con excepción de la pared celular, la práctica totalidad del resto de las dianas de los antimicrobianos se encuentran también en células eucariotas. Las diferencias estructurales entre las bacterias y las células superiores hacen que la afinidad de los antimicrobianos de interés clínico por las dianas procarióticas sea mucho mayor que por las de sus homólogas eucariotas, disminuyendo así el riesgo de efectos adversos. La principales diferencias entre las células bacterianas y las eucariotas incluyen en las primeras2: a) existencia de un único cromosoma en la bacteria, que no está rodeado de membrana nuclear y se halla en contacto directo con el citoplasma (por tanto, muy accesible a los antibióticos que actúan sobre la síntesis de ADN); b) presencia de ribosomas del tipo 70S, y c) presencia de una pared celular con peptidoglicano (excepto en Mycoplasma spp.), estructura que confiere forma y rigidez a la bacteria.
Para que los antimicrobianos alcancen su diana deben atravesar la cubierta bacteriana, salvo cuando la diana es la propia envoltura externa de los gramnegativos. Las bacterias gramnegativas ofrecen mayor resistencia que las grampositivas a la entrada de antimicrobianos, pues poseen una membrana celular externa, que rodea la capa de peptidoglucano. Esa membrana es una bicapa de lipídica que, a diferencia de las membranas eucariotas, contiene lipolisacárido, y desempeña un importante papel de barrera frente a determinados antimicrobianos3. En la misma existen un gran número de proteínas, que representan en torno al 40% de su peso total, entre las cuales se encuentran las porinas, proteínas triméricas o monoméricas que forman conductos o poros hidrófilos que permiten el acceso al peptidoglucano. A través de estos poros difunden de forma pasiva pequeñas moléculas hidrofílicas (menores de 600 Da), pero se impide el paso de otras mayores, por ejemplo los glucopéptidos (peso molecular >1.000 Da). Por el contrario, los antibióticos más lipofilicos difunden a través de la bicapa lipídica, y algunos utilizan un mecanismo de transporte con gasto de energía. En las bacterias grampositivas, que carecen de membrana externa, se estima que el límite de exclusión es de 100 kDa, mucho mayor que el tamaño de la mayoría de los antimicrobianos.
Ya en el interior del microorganismo los antimicrobianos deben evitar su hidrólisis o su transformación en un producto inactivo y reconocer de forma efectiva una diana antes de que algún sistema de expulsión lo lance de nuevo fuera de la bacteria.
Desde el punto de vista molecular, los antimicrobianos de uso clínico ejercen su acción en algunas de las siguientes estructuras o funciones bacterianas: inhibiendo la síntesis de la pared bacteriana, alterando la integridad de la membrana citoplásmica, impidiendo la síntesis proteica o bloqueando la síntesis o las funciones de ácidos nucleicos. Hay también otros antimicrobianos cuya función es proteger otros compuestos de las enzimas hidrolíticas bacterianas, como es el caso de los inhibidores de β-lactamasas4,5.
Atendiendo a su efecto antibacteriano, los antimicrobianos se han clasificado tradicionalmente en bactericidas (ejercen una acción letal para la bacteria) o bacteriostáticos (sólo inhiben transitoriamente el crecimiento bacteriano). Los límites de ambos conceptos se consideran en la actualidad un tanto difusos, como ya se recoge en otro número de EIMC6. Cada grupo de antibióticos actúa preferentemente de una forma u otra, aunque un mismo antibiótico puede comportarse como bactericida o bacteriostático, dependiendo de la concentración que alcance en la diana, o de su afinidad por la diana de un determinado microorganismo. En general, son bactericidas los antimicrobianos que actúan inhibiendo la síntesis de la pared, alterando la membrana citoplásmica o interfiriendo con algunos aspectos del metabolismo del ADN, y bacteriostáticos los que inhiben la síntesis proteica, excepto los aminoglucósidos.
Atendiendo a su mecanismo de acción y estructura química, los principales grupos de antimicrobianos de interés clínico y sus principales representantes se recogen en la tabla 1.
Antimicrobianos que inhiben la síntesis de la pared bacteriana
La pared celular protege la integridad anatomofisiológica de la bacteria y soporta su gran presión osmótica interna (mayor en las bacterias grampositivas). La ausencia de esta estructura condicionaría la destrucción del microorganismo, inducida por el elevado gradiente de osmolaridad que suele existir entre el medio y el citoplasma bacteriano7. Los antibióticos que inhiben la síntesis de la pared necesitan para ejercer su acción que la bacteria se halle en crecimiento activo, y para su acción bactericida requieren que el medio en que se encuentre la bacteria sea isotónico o hipotónico, lo que favorece el estallido celular cuando la pared celular se pierde o se desestructura. Suelen ser más activos sobre las bacterias grampositivas por su mayor riqueza en peptidoglucano. En general, son poco tóxicos por actuar selectivamente en una estructura que no está presente en las células humanas.
La síntesis de la pared celular se desarrolla en 3 etapas, sobre cada una de las cuales pueden actuar diferentes compuestos: la etapa citoplásmica, donde se sintetizan los precursores del peptidoglucano; el transporte a través de la membrana citoplásmica, y la organización final de la estructura del peptidoglucano, que se desarrolla en la parte más externa de la pared.
Inhibidores de la fase citoplásmica
En el citoplasma bacteriano se sintetizan los precursores del peptidoglucano a partir de diferentes elementos: uridindifosfato-N-acetil-glucosamina (UDP-NAG), ácido fosfoenolpirúvico, uridintrifosfato (UTP) y NADH, a partir de los cuales se forma el ácido uridindifosfato-N-acetilmurámico (UDP-NAM). Después se unen al azúcar una cadena de aminoácidos (frecuentemente 5) en la que se alternan las formas L y D y en la que los dos últimos conforman el dipéptido D-alanin-D-alanina.
En esta etapa de síntesis de precursores de peptidoglucano actúan la fosfomicina y la cicloserina.
1. Fosfomicina. Actúa inhibiendo la piruviltransferasa, enzima causante de la adición del fosfoenolpiruvato a la molécula de UDP-NAG para formar el precursor UDP-NAM. Esta reacción se inhibe porque la fosfomicina, que es un análogo estructural del fosfoenolpiruvato, se une covalentemente con la enzima. La fosfomicina atraviesa la membrana externa mediante las porinas; debido a su pequeño tamaño pasa la barrera de peptidoglicano sin dificultad y finalmente atraviesa la membrana citoplásmica a través de sistemas de transporte activo, uno de los cuales es de expresión inducible, que se favorece en presencia de glucosa-6-fosfato. Por eso, a los medios o discos para estudiar la sensibilidad de las bacterias a la fosfomicina debe añadirse glucosa-6-fosfato8.
Es un antibiótico de amplio espectro que incluye bacilos gramnegativos y grampositivos y Staphylococcus spp. (excepto S. saprophyticus y S. capitis).
2. Cicloserina. Actúa sobre la base de su analogía estructural con la D-alanina, inhibiendo competitivamente la actividad de la L-alanina-racemasa (transforma L-ala en D-ala) y la D-alanin-D-alanina-sintetasa (forma dímeros de D-ala). Por su elevada toxicidad sólo se usa como fármaco antimicobacteriano de segunda línea.
Inhibidores de la fase de transporte de precursores
En esta fase, que se desarrolla en la membrana citoplásmica, un transportador lipídico tomará a su cargo el precursor formado en el citoplasma y lo hará atravesar la membrana citoplásmica. Se trata de un fosfolípido de 55 átomos de carbono, el undecaprenilfosfato. También en la membrana citoplásmica, termina de formarse el precursor mediante la adición de una molécula de N-acetilglucosamina, que se enlaza al átomo C1 del ácido murámico, formándose así un polímero lineal de peptidoglucano constituido por unidades de NAG y NAM-pentapétido. Una vez que este precursor disacárido-pentapéptido es transferido a un lugar aceptor en la pared preexistente, el transportador queda pirofosforilado y se separa, y debe presentar una defosforilación para convertirse en su forma monofosfato activa, que puede transportar ya nuevos precursores a la capa de peptidoglucano.
Bacitracina
Este antimicrobiano se une al transportador y bloquea su defosforilación, e impide que pueda utilizarse de nuevo en el transporte de los polímeros lineales de disacárido-pentapéptido a través de la membrana citoplásmica, hasta la pared en formación. Este antibiótico es activo contra cocos grampositivos (excepto estreptococos del grupo B), Neisseria spp. y de forma variable C. difficile.
Mureidomicinas
Son un nuevo grupo de antimicrobianos producidos por Streptomyces flavidivirens, que por su analogía estructural con el precursor disacárido pentapéptido, se unen competitivamente con el transportador lipídico, bloqueando el transporte de los precursores a través de la membrana citoplásmica. No existen derivados de uso clínico.
Inhibidores de la organización estructural del peptidoglucano
En esta etapa, los precursores de peptidoglucano se ensamblan con la ayuda de enzimas situados en su superficie conocidos como proteínas fijadoras de penicilina (penicillin binding proteins [PBP]). En esta etapa tienen su acción los glucopéptidos y los β-lactámicos.
Glucopéptidos
Los glucopéptidos (vancomicina y teicoplanina) actúan en un paso previo al de los β-lactámicos. Impiden la transferencia del disacárido pentapéptido, unido al transportador lipídico de la membrana citoplásmica, al aceptor de la pared celular. Esto se debe a que estos compuestos recubren el extremo D-alanin-D-alanina del disacárido-pentapéptido, evitando así la acción de las glucosiltransferasas y transpeptidasas, y en consecuencia evitando la elongación del peptidoglucano9.
El gran tamaño de estas moléculas impide su paso a través de la pared de las bacterias gramnegativas, de forma que sólo resultan activas frente a grampositivos (incluyendo con gran frecuencia cepas multirresistentes). Aunque son bactericidas frente a Staphylococcus spp., sólo tienen actividad bacteriostática frente a Enterococcus spp.
β-lactámicos
Representan el grupo más numeroso y de mayor uso en clínica. Su nombre deriva de la presencia de un anillo lactámico en su estructura, con un oxígeno en posición β con respecto a un nitrógeno. En función de los radicales que se unen a este anillo se distinguen varios subgrupos, de los que los más importantes son: penicilinas, cefalosporinas, monobactamas y carbapenemas10.
Los β-lactámicos son compuestos bactericidas que inhiben las fases finales de la síntesis del peptidoglucano, en la que intervienen activamente las ya citadas encimas PBP. Las PBP tienen actividad transpeptidasa, transglucosilasa y carboxipetidasa, por lo que pueden entrelazar los componentes del peptidoglucano11. Los β-lactámicos bloquean estas enzimas porque el anillo β-lactámico tiene una estructura espacial similar a la del residuo acil-D-alanin-D-alanina de las cadenas del peptidoglicano, que es el sustrato natural de las PBP.
Las bacterias poseen varias PBP, cuyas funciones difieren unas de otras. Por esta razón, las consecuencias de su bloqueo también son distintas. Las PBP-1a y PBP-1b actúan como transpeptidasas causantes de la elongación, y su bloqueo provoca la formación de esferoplastos que rápidamente se lisan. La PBP-2 determina la forma bacteriana, y su inhibición da lugar a formas ovoideas que se lisan fácilmente. La PBP-3 interviene en la división bacteriana, y su bloqueo provoca la aparición de formas filamentosas sin septos. Las PBP-4, PBP-5 y PBP-6 tienen actividad carboxipeptidasa, e intervienen en la liberación del quinto aminoácido del pentapéptido, necesaria para la polimerización del peptidoglucano.
Pese a tener el mismo mecanismo de acción, hay diferencias en la actividad de los diferentes β-lactámicos, y ello se debe principalmente a 3 factores: rapidez en la difusión de los antibióticos al espacio periplásmico, resistencia a las β-lactamasas, capacidad para escapar a los sistemas de expulsión activa y afinidad variable por las distintas PBP. Así, un β-lactámico que difunda rápidamente, tenga una gran estabilidad frente a las β-lactamasas, no sea sustrato de las bombas de expulsión y tenga una alta afinidad por las PBP más críticas, será un antibiótico de gran actividad como es el caso de las cefalosporinas de cuarta generación y los carbapénemes.
La acción bactericida de los β-lactámicos no está relacionada directamente con la inhibición de la síntesis de peptidoglucano impidiendo su crecimiento (efecto bacteriostático), sino con la activación de un sistema de enzimas líticos (autolisinas) que, a la inversa de las PBP, están implicadas en la degradación del peptidoglucano12. El peptidoglucano está en continua renovación, resultante del equilibrio entre los procesos de síntesis (PBP) e hidrólisis (autolisinas a bajo nivel de actividad); la rotura de este equilibrio por la acción de los β-lactámicos (inhibición de las PBP y activación de las autolisinas) provoca la muerte bacteriana.
Las bacterias que carecen de autolisinas son inhibidas pero no destruidas, por lo que se dice que son tolerantes. En clínica, se define el fenómeno de tolerancia como la necesidad de una concentración al menos 32 veces mayor a la CMI para que un antimicrobiano destruya una cepa bacteriana13.
El espectro de los β-lactámicos abarca las bacterias grampositivos, gramnegativas y espiroquetas. No son activos frente a micoplasmas por carecer de pared celular, ni frente a bacterias intracelulares como Chlamydia spp. y Rickettsia spp.
Antimicrobianos que bloquean mecanismos de resistencia
Los más importantes son los inhibidores de β-lactamasas de serina, que incluyen ácido clavulánico, sulbactam y tazobactam14. Carecen (habitualmente) de acción antibacteriana intrínseca de verdadera importancia clínica, pero se unen irreversiblemente a algunas β-lactamasas, protegiendo de su acción a los antibióticos β-lactámicos. El sulbactam, además, es activo frente a A. baumannii.
Aunque se conocen sustancias que bloquean in vitro las bombas de expulsión activa o las enzimas modificadoras de aminoglucósidos, ninguna de ellas ha podido introducirse en terapéutica.
Antibióticos activos en la membrana citoplásmica
La membrana citoplásmica es vital para todas las células, ya que interviene activamente en los procesos de difusión y transporte activo, y de esta forma controla la composición del medio interno celular. Las sustancias que alteran esta estructura modifican la permeabilidad, y provocan la salida de iones potasio, elementos esenciales para la vida bacteriana, o la entrada de otros que a altas concentraciones alteran el metabolismo bacteriano normal.
Los antimicrobianos que actúan en esta estructura se comportan como bactericidas, incluso en bacterias en reposo, y pueden tener alta toxicidad sobre las células humanas, al compartir algunos componentes de la membrana citoplásmica.
A este grupo pertenecen las polimixinas, los lipopétidos, los antibióticos poliénicos (activos frente a hongos) y 2 grupos de escaso interés clínico (ionóforos y formadores de poros).
Polimixinas
Son antibióticos polipeptídicos, cíclicos y policatiónicos, con una cadena de ácido graso unido al péptido y se comportan como detergentes catiónicos. Tiene una parte hidrofílica (el péptido) con alta carga positiva que por atracción electrostática se une a la superficie de la membrana, cuya carga neta es negativa. Por otro lado, el extremo lipofílico (la cadena lateral de ácido graso) por interacciones hidrofóbicas se une a los fosfolípidos de la membrana. Como resultado se desorganiza la estructura de la membrana y aumenta su permeabilidad, con la pérdida de metabolitos esenciales15. La mayor presencia de fosfolípidos en la membrana de las bacterias gramnegativas hace que éstas sean más sensibles que las grampositivas a la acción de estos agentes.
Son activos exclusivamente frente a bacilos gramnegativos aerobios, incluidos P. aeruginosa y A. baumannii multirresistentes. No son activos frente a microorganismos anaerobios, Proteus spp., Providencia spp., Serratia spp., Neisseria spp. y B. cepacia.
Daptomicina
La daptomicina es un lipopéptido cíclico de reciente introducción en clínica que posee una gran actividad frente a bacterias grampositivas. Actúa en la membrana citoplásmica de las bacterias grampositivas, sin entrar en la célula, y se produce una rápida despolarización de la membrana con alteración del potencial eléctrico y salida de iones potasio exterior. Como consecuencia de ello, se produce un bloqueo de la síntesis proteica y de ácidos nucleicos, que provoca la muerte bacteriana16. La daptomicina es una gran molécula cíclica peptídica (parte hidrofílica), al que se une una cadena lateral de ácido graso (parte lipofílica). Se sabe que su actividad antibacteriana depende de la presencia de iones de calcio que es óptima a las concentraciones normales presentes en suero (50 mg/l). Probablemente, el calcio favorece la unión de la parte lipofílica de la molécula de daptomicina a la membrana citoplásmica, donde la estructura de la daptomicina presentará cambios conformacionales que provocará su inserción en la membrana citoplásmica. La unión de varias moléculas en la membrana forma canales por los que saldrán los iones potasio. El resultado es una potente actividad bactericida, con efecto postantibiótico.
Su espectro antimicrobiano se reduce a las bacterias grampositivas, ya que no puede atravesar la pared de los gramnegativos.
Ionóforos y formadores de poros
Los ionóforos son antibióticos polipeptídicos cíclicos como la valinomicina o las tirocidinas A y B. Estos compuestos tiene una estructura circular peculiar, es hidrofóbica en el exterior e hidrofílica o polar en el interior. Los ionóforos incorporan cationes monovalentes en su interior, y les permite cruzar la bicapa lipídica. La penetración elevada de potasio altera el potencial eléctrico y el gradiente químico existente en la membrana, alterando su función.
Los antibióticos formadores de poros incluyen las gramicidinas, que a diferencia de los ionóforos, son cadenas lineales de aminoácidos (polipéptidos acíclicos) con un mecanismo de acción distinto. Varias moléculas de gramicidina se acomodan una sobre otra, enroscándose y formando un túnel que atraviesa la membrana, constituyendo un poro que permite el paso selectivo de moléculas según su tamaño y características. La gramicidina se encuentra en formulaciones tópicas para tratamiento de conjuntivitis bacterianas, y resulta efectiva frente a bacterias grampositivas. Es un agente hemolítico potente, y su alta toxicidad lo descarta para uso sistémico. Además se inactiva en suero y líquidos orgánicos.
Antibióticos inhibidores de la síntesis proteica
La síntesis proteica es uno de los procesos más frecuentemente afectados por la acción de los antimicrobianos, y su inhibición selectiva es posible gracias a las diferencias estructurales entre los ribosomas bacterianos y eucariotas. Los ribosomas bacterianos están formados por dos subunidades (30S y 50S), que contienen ARN ribosómico (ARNr 16S en la subunidad 30S, y ARNr 5S y ARNr 23S en la subunidad 50S) y diversas proteínas llamadas S (small o pequeña, en la subunidad 30S) o L (large o grande, en la subunidad 50S). En esta estructura diferentes componentes pueden ser lugares de unión para los antimicrobianos (p. ej., determinados nucleótidos para las oxazolidinonas, algunas proteínas S para las tetraciclinas o proteínas L para el cloranfenicol).
La mayoría de los antibióticos de este grupo tienen actividad bacteriostática, aunque los aminoglucósidos se comportan como bactericidas. La acción bactericida o bacteriostática también va a depender de las concentraciones del antimicrobiano, y del microorganismo afectado.
La síntesis proteica se desarrolla en diferentes fases17, en las cuales actúan diferentes antimicrobianos, como se explica a continuación.
Inhibidores de la fase de activación
Los aminoácidos son transportados a la cadena peptídica en formación en el ribosoma, por moléculas de ARN de transferencia (ARNt) que se unirán al ARNm codificante de la proteína en formación. Para ello, cada aminoácido se une con su ARNt específico mediante una enzima también específica de aminoácido (aminoacil ARNt sintetasa). En las bacterias, el primer aminoácido de la cadena peptídica es la metionina, es decir, la síntesis proteica se inicia con la formación del complejo formilmetionil-ARNt que reconocerá el codón de iniciación AUG del ARNm (adenosina-uracilo-guanosina).
Mupirocina
Antibiótico bacteriostático obtenido de especies de Pseudomonas spp., que inhibe competitivamente la enzima isoleucil-ARNt sintetasa, con lo cual no puede incorporarse el aminoácido isoleucina al péptido en formación y la síntesis de proteínas se interrumpe18.
Su acción es especialmente potente frente a grampositivos, aunque debido a las altas concentraciones que se alcanzan en piel y mucosa nasal tras su aplicación tópica, puede tener también alguna actividad frente a microorganismos gramnegativos. Se usa fundamentalmente en el tratamiento tópico de infecciones cutáneas o para erradicación del estado de portador de S. aureus.
Inhibidores del inicio de la síntesis proteica
El ARNm dispone de un codón específico para la fijación del ARNt que porta el aminoácido formilmetionina. Ambos se unen en la subunidad 30S, y posteriormente a la subunidad 50S, y constituye el complejo de iniciación de la síntesis de proteínas. En este complejo hay 2 sitios activos, el locus A, en el que se fijan los aminoacil-ARNt, y el locus P, donde se engarza el péptido en formación y donde se ubicará el formilmetionil-ARNt que inicia la cadena peptídica. En esta fase de inicio de la síntesis actúan las oxazolidinonas y los aminoglucósidos.
Oxazolidinonas
Representan una de las últimas familias de antimicrobianos incorporadas a la práctica clínica. Son compuestos obtenidos por síntesis, y su representante en uso es el linezolid19. Las oxazolidinonas inhiben la síntesis proteica e impiden la formación del complejo de iniciación 70S, formado por formilmetionil-ARNt, ARNm, diversas proteínas y las subunidades ribosómicos 30S y 50S. El linezolid se fija a la subunidad ribosómica 50S, en el centro peptidiltransferasa dentro del ARN ribosómico 23S (dominio V), distorsiona así el punto de unión del formilmetionil-ARNt y evita, por tanto, la formación del complejo de iniciación. Esta familia de antibióticos tiene un mecanismo de acción singular, y al actuar en una diana distinta no hay resistencia cruzada con otros antibióticos que también inhiben la síntesis proteica.
El linezolid es bacteriostático frente a bacterias grampositivas (incluidas cepas multirresistentes de S. aureus y Enterococcus spp.) y carece de actividad frente a la práctica totalidad de las bacterias gramnegativas.
Aminoglucósidos
Son compuestos naturales obtenidos de actinomicetos del suelo o productos semisintéticos derivados de ellos. Poseen un anillo aminociclitol al que se unen diferentes azúcares.
Aunque la diana primaria de actuación de los aminoglucósidos está en los ribosomas y sus procesos de síntesis proteica, su actividad sobre las bacterias no se entiende sin conocer los fenómenos que se producen en la membrana. Los aminoglucósidos son moléculas muy cargadas positivamente, lo que les permite concentrarse en torno a las bacterias por atracción de las cargas negativas de la superficie bacteriana, aportadas por los grupos fosfatos de los fosfolípidos de la membrana externa de las bacterias gramnegativas y de los ácidos teióicos unidos al peptidoglucano de las grampositivas. En consecuencia, desplazan los iones de magnesio y calcio que se enlazan a las moléculas de lipopolisacáridos adyacentes; este proceso desestructura la membrana externa y permite al paso de los aminoglucósidos. Una vez pasada fácilmente la barrera de peptidoglucano (grampositivos y gramnegativos), vuelve a concentrarse en torno a la membrana citoplásmica. La difusión a través de esta membrana ocurre en 2 fases: una inicial lenta y otra posterior rápida; ambas dependientes de la energía generada por el transporte de electrones que implica la participación de sistemas enzimáticos del metabolismo aerobio, que crea un gradiente eléctrico a ambos lados de la membrana20. Este hecho explica la ineficacia de estos compuestos frente a microorganismos anaerobios. La presencia de iones de magnesio y calcio en el medio y las situaciones que disminuyen el potencial transmembrana (pH ácido, ambiente anaerobio o hiperosmolaridad), reducen la difusión del aminoglucósidos al interior de la bacteria y aumentan las CIM de forma importante. Una vez que los aminoglucósidos han empezado a actuar en los ribosomas, comienzan a producirse muchos errores en la lectura del ARNm, que darán como resultado proteínas anómalas que se unirán a la membrana, deteriorando su integridad y acelerando la difusión de más moléculas de aminoglucósido (fase rápida). En consecuencia, una gran cantidad de aminoglucósidos alcanza los ribosomas, que llegan a bloquearse, y se detienen irreversiblemente la síntesis de proteínas.
En el ribosoma, los aminoglucósidos tienen su acción principalmente en la subunidad 30S, donde se unen a diferentes proteínas S y al ARN 16S. Bloquean la actividad normal del complejo de iniciación, impiden el inicio de la síntesis y provocan también una lectura errónea del ARNm.
Los aminoglucósidos tienen un efecto bactericida dependiente de su concentración y poseen un importante efecto postantibiótico21, es decir que una breve exposición de la bacteria a estos compuestos induce una supresión de su crecimiento, aun cuando el antimicrobiano no alcance ya concentraciones que inhiban o maten al microorganismo. Son activos frente a un amplio número de especies bacterianas, especialmente frente a microorganismos gramnegativos aerobios.
Inhibidores de la fijación del aminoacil-ARNt al ribososma
Una vez iniciada la síntesis proteica, el proceso continúa con la incorporación de nuevos aminoácidos al locus A, donde reconocerán los codones internos del ARNm a través de los nucleótidos complementarios del ARNt que porta el aminoácido. Esta fase se ve bloqueada por antibióticos bacteriostáticos como las tetraciclinas y sus derivadas, las glicilciclinas.
Tetraciclinas
Son moléculas naturales o semisintéticas con un núcleo hidronaftaceno, que contiene cuatro anillos fundidos al que se pueden unir distintos radicales que darán lugar a las diferentes tetraciclinas. Penetran en el citoplasma bacteriano por un proceso dependiente de energía y se unen de forma reversible a la subunidad 30S del ribosoma (proteínas S7, S14, S19), bloqueando el acceso de los complejos aminoacil-ARN-t, e impidiendo la continuación de la síntesis proteica22. Estos compuestos pueden también unirse (aunque menos selectivamente) a los ribosomas 80S de las células humanas y efectuar la misma acción; sin embargo, carecen de los medios de transporte específicos de la membrana bacteriana.
El compuesto más usado es la doxiciclina, y en España también están disponibles minociclina, oxitetraciclina y tetraciclina.
Son el mejor ejemplo de lo que se denomina antibióticos de amplio espectro, con actividad tanto para grampositivos como frente a gramnegativos, pero esta actividad depende de los grados de resistencia observados en cada especie, que son muy variables. Son también activas frente a micobacterias atípicas, rickettsias, micoplasmas, clamidias, espiroquetas, Coxiella burnetii y algunos protozoos.
Glicilciclinas
Son compuestos sintéticos derivados de las tetraciclinas, de las cuales está comercializada la tigeciclina, un derivado de la minociclina. Poseen el mismo mecanismo de acción aunque se unen al ribosoma con una afinidad 5 veces superior que la minociclina23. Además, las glicilciclinas se fijan a la membrana citoplásmica y alteran su permeabilidad.
La tigeciclina posee el amplio espectro propio de las tetraciclinas, pero es más potente e incluso activa contra bacterias con modificaciones ribosómicas resistentes a las mismas, incluyendo enterococos resistentes a glucopéptidos, S. aureus resistente a meticilina, S. pneumoniae multirresistentes y diversas bacterias gramnegativas resistentes a otros compuestos.
Inhibidores de la elongación
Una vez que el ARNt que porta un aminoácido se ha fijado al locus A, el centro peptidiltransferasa, situado en la subunidad 50S, cataliza la unión entre el aminoácido incorporado y el último aminoácido del péptido en formación (locus P), proceso denominado transpeptidación, que puede estar bloqueado por el cloranfenicol y las lincosamidas.
Una vez formado el enlace peptídico, el ARNt fijado al locus P se libera y se separa de su aminoácido correspondiente, quedando libre el locus P. Seguidamente se produce la translocación del peptidil-ARNt del locus A al locus P, desplazándose la subunidad 30S un codón a lo largo del ARNm. De esta manera queda libre el locus A, y preparado para recibir un nuevo ARNt con su correspondiente aminoácido. Este proceso, que conlleva gasto de energía, requiere la participación clave del factor de elongación G, que puede estar bloqueado por el ácido fusídico. El péptido en formación va pasando a través de un canal peptídico en la subunidad 50S y emerge por la parte posterior del ribosoma, y este proceso puede estar bloqueado por los antibióticos del grupo MLSB (macrólidos, lincosamidas y estreptograminas del grupo B) y por los cetólidos. La mayoría de los aminoglucósidos también ejercen su acción interfiriendo con la fase de elongación peptídica.
Todos los antimicrobianos que inhiben la transpeptidación y la translocación actúan sobre la subunidad ribosómica 50S.
Anfenicoles
El cloranfenicol y su derivado, el tiamfenicol, son antibióticos bacteriostáticos que bloquean la síntesis proteica bacteriana uniéndose reversiblemente a la proteína L16 localizada en la subunidad 50S. Esta proteína es la que media la fijación del ARNt a la enzima peptidiltransferasa, y por tanto, su bloqueo por el cloranfenicol evita la formación de los enlaces peptídicos.
Tiene un amplio espectro de actividad contra microorganismos grampositivos, gramnegativos y anaerobios. Su espectro incluye a neisserias, Haemophilus spp, clamidias, rickettsias, micoplasmas y espiroquetas.
Lincosamidas
La principal lincosamida es clindamicina, un derivado semisintético de la lincomicina, que es un aminoácido unido a un aminoazúcar. Generalmente bacteriostáticos, pueden ser bactericidas dependiendo de su concentración y del microorganismo considerado.
Actúa inhibiendo la síntesis proteica tras unirse reversiblemente a la subunidad 50S del ribosoma, en un lugar próximo al del cloranfenicol o los macrólidos, impidiendo la acción de la peptidiltransferasa24.
Es activa frente a bacterias grampositivas, excepto enterococos y microorganismos anaerobios, incluido el grupo de B. fragilis. También es activa frente a algunos protozoos como Plasmodium spp. o Toxoplasma gondii. Al igual que los macrólidos y las estreptograminas, no son activas frente a enterobacterias, Pseudomonas spp. u otros gramnegativos aerobios, probablemente porque no pueden atravesar la pared bacteriana.
Macrólidos y cetólidos
Forman un grupo de antimicrobianos que se caracteriza por la presencia de un anillo lactónico macrocíclico al que unen uno o varios azúcares. La eritromicina fue el primer macrólido utilizado en clínica, a partir del cual se introdujeron modificaciones en su estructura química que dieron lugar a derivados semisintéticos con mejores propiedades farmacocinéticas aunque, salvo excepciones, no presentaban mejorías en su actividad antimicrobiana. Dependiendo del número de elementos contenidos en el anillo, se clasifican en: macrólidos de 14 átomos de carbono como eritromicina, claritromicina, roxitromicina. etc.; macrólidos de 15 átomos de carbono como la azitromicina, en la que se incorpora un átomo de nitrógenos entre los carbonos 9 y 10 que da lugar a una estructura nueva conocida como azálido; macrólidos de 16 átomos de carbono como espiramicina, josamicina, midecamicina, etc. Los cetólidos, como la telitromicina, son un grupo nuevo de antibióticos derivados de la eritomicina, en los que el azúcar unido al carbono 3 se sustituye con un grupo cetónico.
Se unen de forma reversible al dominio V del centro peptidiltransferasa, en el ARNr 23S de la subunidad 50S del ribosoma, interfiriendo así el proceso de elongación de la síntesis proteica. Además los cetólidos interacciona también con el dominio II del ARNr 23S por lo que la afinidad de los cetólidos por el ribosoma es mucho mayor que el resto de los macrólidos. Estos lugares de unión se sitúan en el orificio de entrada al túnel ribosómico por donde sale la proteína en formación, de manera que al unirse los macrólidos o los cetólidos, se bloquea este canal, impidiendo estéricamente el crecimiento del péptido25. También se ha descrito en los cetólidos, una inhibición de la formación de los ribosomas 50S al evitar el ensamblaje de los ARNr 5S y 23S con las riboproteínas, con lo que se impide el inicio de la síntesis26.
Son generalmente considerados como bacteriostáticos, aunque a altas concentraciones y con un bajo inóculo pueden mostrar acción bactericida especialmente en Streptococcus spp.
Son activos frente a bacterias grampositivas (incluyendo actinomicetos e incluso micobacterias), Bordetella pertussis, Haemophilus ducreyi, Moraxella spp, Neisseria spp., Campylobacter spp., Helicobacter pylori (claritromicina), treponemas, borrelias, Legionella spp., micoplasmas, clamidias y ricketsias. La azitromicina es algo menos activa que la eritromicina frente a microorganismos grampositivos, pero es más activa frente a gramnegativos. Apenas son activos frente a enterobacterias y P. aeruginosa, aunque parecen ser útiles (sobre todo, azitromicina) para el tratamiento de infecciones respiratorias crónicas por P. aeruginosa, tal vez por propiedades antiinflamatorias o inmunomoduladores o por inhibición de la síntesis de alginato, componente de los biofilms.
Estreptograminas
También llamadas sinerginas, forman un grupo de antimicrobianos con un estructura compleja constituida por una macrolactona (estreptogramina grupo A) y un polipéptido cíclico (estreptogramina grupo B)27. Ambos compuestos actúan sinérgicamente de forma bactericida, bloqueando la acción de la peptidiltransferasa en diferentes puntos. Su principal representante es la asociación quinupristina-dalfopristina en proporción 3:7, con actividad fundamentalmente frente a bacterias grampositivas (excepto E. faecalis) y también frente a algunas bacterias fastidiosas (Moraxella spp., Neisseria spp., Mycoplasma spp., L. pneumophila) y algunos anaerobios (Prevotella y Porphyromonas spp.). Las estreptograminas A (dalfopristina) se unen al ARNr 23S (subunidad 50S), y modifican la estructura terciaria de ciertas proteínas ribosómicas, de manera que aumenta la afinidad por las estreptograminas del grupo B (quinupristina). Este hecho explica su acción bactericida cuando actúan juntos, ya que por separado son sólo bacteriostáticos.
Ácido fusídico
Es un antibiótico de estructura esteroide que se une al complejo causante de la translocación formado por el factor de elongación G, GDP y el ribosoma. Al unirse al complejo, lo estabiliza e impide la liberación del factor de elongación G para una nueva translocación.
Puede comportarse como bacteriostático o bactericida según la concentración y el microorganismo. Es de espectro reducido a los microorganismos grampositivos como S. aureus, S. epidermidis, Clostridium spp. y Corynebacterium spp., aunque también puede ser activo frente a meningococos, gonococos y algunos protozoos como Giardia lamblia y Plasmodium falciparum.
Antibióticos que actúan en el metabolismo o la estructura de los ácidos nucleicos
El genoma bacteriano contiene información para la síntesis de proteínas que se transmite a través del ARN mensajero producido a partir del molde de ADN (transcripción), y para la síntesis de ARN ribosómico que formará parte de los ribosomas bacterianos. La información del ADN debe duplicarse (replicación) cuando la bacteria se divide, para transmitir esta información a la descendencia. La replicación y la transcripción del ADN se realizan en varias fases con la participación de diferentes enzimas y sustratos, además del ADN molde, que constituyen dianas para la acción de diversos antibióticos.
Dentro de este grupo incluimos las rifamicinas y las quinolonas que actúan en enzimas que participan en los procesos de transcripción y replicación, y los nitroimidazoles y nitrofuranos que actúan directamente sobre el ADN, dañándolo. Por lo general, los antibióticos de este grupo no son particularmente selectivos en su acción y comportan cierta toxicidad para las células eucarióticas. La mayoría de los antibióticos que actúan sobre el ADN son bactericidas rápidos y normalmente independientes del inóculo y de la fase de crecimiento bacteriano.
Rifamicinas
Inhiben la síntesis de ARN ribosómico y mensajero al bloquear la subunidad beta de la ARN polimerasa ADN-dependiente bacteriana codificada por el gen rpoB28. Impide el inicio del proceso de transcripción, pero carece de efecto antimicrobiano si la transcripción ya se ha iniciado.
La rifampicina, derivado semisintético de la rifamicina B, es el antibiótico representativo de este grupo y tiene actividad bactericida frente a microorganismos grampositivos, Neisseria spp., Chlamydia spp. y Mycobacterium spp.
Quinolonas
El cromosoma bacteriano está constituido por una doble cadena de ADN que es 1.000 veces más largo que la propia bacteria, por lo que se encuentra muy plegado sobre sí mismo en una forma superenrollada. Esta configuración no es accesible para que pueda realizarse la replicación y transcripción del ADN bacteriano, por lo que debe desenrollarse. Las topoisomerasas son las enzimas encargadas del superenrollamiento y desenrollamiento del ADN, así como del corte, unión y separación de las hebras de ADN, necesarias para los procesos de síntesis de ADN y de partición del cromosoma a las células hijas cuando la bacteria se divide. Las quinolonas ejercen su acción bloqueando las topoisomerasas II (ADN-girasa) y IV29. Ambas son enzimas tetraméricas compuestas por dos subunidades A y 2 subunidades B, codificadas respectivamente por los genes gyrA y gyrB en el caso de la ADN-girasa, y parC y parE en el caso de la topoisomerasa IV. Las topoisomerasas se acoplan al ADN, provocan un pequeño corte en las hebras de ADN que posteriormente es reparado, y quedan de nuevo libres. Las fluoroquinolonas, se unen al ADN roto y a la topoisomerasa formando un complejo ternario quinolona-ADN-topoisomerasa de forma irreversible, impidiendo que el proceso de transcripción o replicación continúen.
Su acción en las topoisomerasas no explica por sí sola su potente acción bactericida, sino que se debe a fenómenos secundarios mal conocidos30, entre los que la activación del sistema de reparación de mutaciones SOS parece desempeñar un papel importante.
Las quinolonas constituyen en la actualidad, junto a los β-lactámicos, los antibióticos de mayor uso. De forma semejante a lo que ocurre con las cefalosporinas, las quinolonas se han clasificado en generaciones, atendiendo a su espectro de actividad y propiedades farmacocinéticas. Las quinolonas de primera generación (ácido nalidíxico) tienen un espectro limitado a bacilos gramnegativos y sólo se utilizan para infecciones de tracto urinario. La introducción de un átomo de flúor ha permitido la síntesis de nuevas generaciones (fluoroquinolonas) con mejor actividad farmacocinética. Las de segunda generación (norfloxacino) son mucho más activas frente a gramnegativos, tienen alguna actividad frente a grampositivos, pero no frente a anaerobios. Las de tercera generación (ciprofloxacino, levofloxacino) tienen mejor actividad frente a grampositivos y organismos fastidiosos y por sus propiedades farmacocinéticas permiten su empleo sistémico. Finalmente, las de cuarta generación (moxifloxacino, gemifloxacino) son muy activas frente a grampositivos y tienen una buena actividad antianaerobia.
Nitroimidazoles
Amplio grupo de compuestos de los cuales metronidazol, tinidazol y ornidazol son los más conocidos. Estos antibióticos penetran fácilmente en el citoplasma por difusión pasiva y allí el grupo NO2 del anillo imidazólico, que se comporta como aceptor de electrones, se reduce por nitroreductasas bacterianas del metabolismo anaerobio, liberándose radicales nitritos que dañan el ADN por oxidación. Tienen actividad frente a Clostridium spp., microorganismos gramnegativos anaerobios y microorganismos microaerofílicos (Helicobacter pylori, Campylobacter spp., Gardnerella vaginalis) y protozoos (tricomonas, giardias, amebas, Balantidium coli).
Nitrofuranos
La nitrofurantoína es el antibiótico representativo de este grupo y se usa como antiséptico urinario. Al igual que los nitroimidazoles, estos compuestos se reducen en el citoplasma bacteriano para generar derivados tóxicos que dañan el ADN por un mecanismo no bien conocido. También parecen interferir con la síntesis proteica bacteriana al unirse al ribosoma 30S bloqueando el reconocimiento del codón-anticodón.
Bloqueo de la síntesis de factores metabólicos
Para obtener determinados elementos esenciales como los aminoácidos o las bases púricas y pirimidínicas de los nucleótidos, se requiere la síntesis de folatos, que algunas bacterias son incapaces de obtener del medio, a diferencia de las células eucariotas. La síntesis de ácido tetrahidrofólico se obtiene a partir de una molécula de pteridina y de ácido paraaminobenzoico (PABA), y mediante la enzima dihidropteroatosintetasa se forma el ácido dihidropteroico. Posteriormente, por adición de ácido glutámico se forma el ácido dihidrofólico (ácido fólico), que reducido por la dihidrofolato reductasa forma el ácido tetrahidrofólico (ácido folínico).
Sulfamidas, diaminopirimidinas
Las sulfamidas son análogos del ácido paraaminobenzoico, y por tanto, compiten por la enzima dihidropteroatosintetasa, impidiendo así la formación de ácido dihidropteroico, precursor del ácido fólico. Estos antibióticos no afectan a las células humanas, que obtienen ácido fólico de la dieta. De este grupo se usa en clínica, sulfametoxazol (asociado a trimetoprima), sulfisoxazol, suladiazina, sulfacetamida, etc.
Las diaminopirimidinas, como la trimetoprima y la pirimetamina, compiten por la enzima dihidrofolatoreductasa que cataliza la conversión de ácido dihidrofólico en ácido tetrahidrofólico. El trimetoprima tiene mucha menos afinidad por la dihidrofolatoreductasa humana, que, sin embargo, puede llegar a afectarse con dosis altas o en pacientes con alteraciones hemáticas preexistentes.
El cotrimoxazol31 es la combinación de trimetoprima y sulfametoxazol en proporción 1:5, y por tanto, actúa en dos etapas de la síntesis de ácido folínico, pudiendo llegar a tener efecto bactericida por la sinergia ente sus 2 componentes.
Otras combinaciones utilizadas son las formadas por pirimetamina y sulfadiazina para el tratamiento de toxoplasmosis, o pirimetamina y sulfadoxina para el paludismo.
Autor para correspondencia.
Luis Martínez-Martínez
Dirección: lmartinez@humv.es
Bibliografía
1. Fleming A. On the antibacterial action of cultures of a Penicillium, with special reference to their use in the isolation of B. inlfuenzae. Brit J Exp Pathol. 1929;10:226-236.
2. Fleming A. Ausina Ruiz V, Prats Pastor G. Principales grupos de seres vivos con capacidad patógena para el hombre. En: Auxina Ruiz V, Moreno Guillén S, directores. Tratado SEIMC de enfermedades infecciosas y microbiología clínica. Madrid: Editorial Médica-Panamericana; 2006. p. 1–18.
3. Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev. 2003;67:593-656.[Medline]
4. Nikaido H. Pascual Hernández A, Martínez-Martínez L, Almirante Gragera B, Miró Meda JM, editores. Actualización en antimicrobianos. Sociedad Española de Enfermedades Infecciosas y Microbiología Clínica. Barcelona: Ediciones Doyma; 2004.
5. García Rodríguez JA. Antimicrobianos en medicina. Prous Science, 2006.
6. Martínez-Martínez L. Muerte bacteriana y heterorresistencia a los antimicrobianos. Enf Infecc Microbiol Clin. 2008;26:481-484.
7. Dover LG, Alderwick LJ, Brown AK, Futterer K, Besra GS. Regulation of cell wall synthesis and growth. Curr Mol Med. 2007;7:247-276.
8. Falagas ME, Giannopoulou KP, Kokolakis GN, Rafailidis PI. Fosfomycin: use beyond urinary tract and gastrointestinal infections. Clin Infect Dis. 2008;46:1069-1077.
9. Allen NE, Nicas TI. Mechanism of action of oritavancin and related glycopeptide antibiotics. FEMS Microbiol Rev. 2003;26:511-532.
10. Asbel LE, Levison ME. Cephalosporins, carbapenems, and monobactams. Infect Dis Clin North Am. 2000;14:435-447.
11. Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev. 2008;32:234-258.[Fe de errores en: FEMS Microbiol Rev. 2008;32:556]
12. Bayles KW. The bactericidal action of penicillin: new clues to an unsolved mystery. Trends Microbiol. 2000;8:274-278.
13. Henriques Normark B, Normark S. Antibiotic tolerance in pneumococci. Clin Microbiol Infect. 2002;8:613-622.
14. Georgopapadakou NH. Beta-lactamase inhibitors: evolving compounds for evolving resistance targets. Expert Opin Investig Drugs. 2004;13:1307-1318.
15. Li J, Nation RL, Turnidge JD, Milne RW, Coulthard K, Rayner CR. Colistin: the re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect Dis. 2006;6:589-601.
16. Kanafani ZA, Corey GR. Daptomycin: a rapidly bactericidal lipopeptide for the treatment of Gram-positive infections. Expert Rev Anti Infect Ther. 2007;5:177-184.
17. Kaczanowska M, Rydén-Aulin M. Ribosome biogenesis and the translation process in Escherichia coli. Microbiol Mol Biol Rev. 2007;71:477-494.
18. Ochsner UA, Sun X, Jarvis T, Critchley I, Janjic N. Aminoacyl-tRNA synthetases: essential and still promising targets for new anti-infective agents. Expert Opin Investig Drugs. 2007;16:573-593.
19. Vara Prasad JV. New oxazolidinones. Curr Opin Microbiol. 2007;10:454-460.
20. Mingeot-Leclercq MP, Glupczynski Y, Tulkens PM. Aminoglycosides: activity and resistance. Antimicrob Agents Chemother. 1999;43:727-737.
21. Lacy MK, Nicolau DP, Nightingale CH, Quintiliani R. The pharmacodynamics of aminoglycosides. Clin Infect Dis. 1998;27:23-27.
22. Shlaes DM. An update on tetracyclines. Curr Opin Investig Drugs. 2006;7:167-171.
23. Zhanel GG, Homenuik K, Nichol K, Noreddin A, Vercaigne L, Embil J. The glycylcyclines: a comparative review with the tetracyclines. Drugs. 2004;64:63-88.
24. Spízek J, Rezanka T. Lincomycin, clindamycin and their applications. Appl Microbiol Biotechnol. 2004;64:455-464.
25. Retsema J, Fu W. Macrolides: structures and microbial targets. Int J Antimicrob Agents. 2001;18:S3-S10.
26. Nilius AM, Ma Z. Ketolides: the future of the macrolides?. Curr Opin Pharmacol. 2002;2:493-500.
27. Blondeau JM, Sanche SE. Quinupristin/dalfopristin. Expert Opin Pharmacother. 2002;3:1341-1364.
28. Villain-Guillot P, Bastide L, Gualtieri M, Leonetti JP. Progress in targeting bacterial transcription. Drug Discov Today. 2007;12:200-208.
29. Hooper DC. Mechanisms of action of antimicrobials: focus on fluoroquinolones. Clin Infect Dis. 2001;32:S9-S15.
30. Drlica K, Malik M, Kerns RJ, Zhao X. Quinolone-mediated bacterial death. Antimicrob Agents Chemother. 2008;52:385-392.
31. Masters PA, O’Bryan TA, Zurlo J, Miller DQ, Joshi N. Trimethoprim-sulfamethoxazole revisited. Arch Intern Med. 2003;163:402-410.
jueves, 23 de septiembre de 2010
Perianal Swabs Detect 80% of Multidrug-Resistant Gram-Negative Infections
Alice Goodman
September 13, 2010 (Boston, Massachusetts) — Perianal swabs failed to detect 1 in 5 cases of multidrug-resistant Gram-negative (MDRGN) bacteria in hospitalized patients with a newly identified MDRGN culture.
Researchers were not able to identify patient factors or types of care that could predict failure of the swab to identify MDRGN bacteria in this study, which was conducted in hospitalized patients known to be infected with MDRGN bacteria. The results were presented here at the 50th Interscience Conference on Antimicrobial Agents and Chemotherapy. A reliable method of detecting MDRGN bacteria in infected but asymptomatic patients has the potential to prevent the spread of MDRGN infection, the authors pointed out.
"We used these swabs in patients who were positive for MDRGN bacteria and we anticipated that we would detect MDRGN bacteria in 100%. Our study suggests that the ability to detect MDRGN bacteria among the generalized hospitalized population may be limited," said lead author Graham M. Snyder, MD, a fellow at Beth Israel Deaconess Hospital in Boston, Massachusetts. Erika D'Agata, MD, and Lara Venkataraman, PhD, also from Beth Israel Deaconess Hospital, were coauthors.
"Future research should be aimed at more accurate methods of surveillance than the swab used in our study and investigate whether there are groups of patients for whom the swab may be more accurate," Dr. Snyder added.
This study used standard agar plates infiltrated with ciprofloxacin or ceftazidime to screen for MDRGN bacteria. Other methods include PCR-based tests for carbapenemase-producing organisms and CHROMagar, he said. Perianal swabs were used because they are easy to perform and more comfortable than full rectal swabs, Dr. Snyder said. In addition to the rectum, potential sites for swabs include the groin, stool, wound, and perineum.
Thirty-five patients agreed to participate in the study, 3 of whom harbored 2 different species of MDRNG bacteria, for a total of 38 distinct MDRNG-patient pairs. The surveillance swab correctly identified 30 of the 38 (78.9%) MDRGN bacteria from clinical isolates. The perianal skin swab identified 1 or both MDRNG clinical isolates in 29 (82.9%) of the 35 patients.
Dr. Snyder gave some potential explanations for the findings. "Failure to detect 20% of MDRNG bacteria could be related to a time lag between identification of the organism initially and using a perianal swab. Or perhaps other sites were colonized and we didn't use a swab on those sites. It is also possible that our methods [microbiological plates] may not have been sensitive enough," he suggested.
"In the big picture, we need to carefully consider the methods we use for active surveillance. We need to think about the site of the swab and the methods we use," Dr. Snyder told Medscape Medical News.
More Optimistic View of Study
"I was impressed by the sensitivity of Dr. Snyder's method, which was around 80% in this study. The study consisted of patients already on antibiotics, which would theoretically have decreased the sensitivity of surveillance swabs to detect the organisms," said Mary-Claire Roghmann, MD, from the University of Maryland in Baltimore.
Dr. Roghmann told Medscape Medical News that she thought that the method of perianal swab testing could have higher sensitivity if performed prior to treatment with antibiotics, as occurred in this study.
"Based on this study, I actually think that the use of perianal swabs is promising," she stated.
Dr. Snyder and Dr. Roghmann have disclosed no relevant financial relationships.
50th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC): Abstract K-317. Presented September 12, 2010.
September 13, 2010 (Boston, Massachusetts) — Perianal swabs failed to detect 1 in 5 cases of multidrug-resistant Gram-negative (MDRGN) bacteria in hospitalized patients with a newly identified MDRGN culture.
Researchers were not able to identify patient factors or types of care that could predict failure of the swab to identify MDRGN bacteria in this study, which was conducted in hospitalized patients known to be infected with MDRGN bacteria. The results were presented here at the 50th Interscience Conference on Antimicrobial Agents and Chemotherapy. A reliable method of detecting MDRGN bacteria in infected but asymptomatic patients has the potential to prevent the spread of MDRGN infection, the authors pointed out.
"We used these swabs in patients who were positive for MDRGN bacteria and we anticipated that we would detect MDRGN bacteria in 100%. Our study suggests that the ability to detect MDRGN bacteria among the generalized hospitalized population may be limited," said lead author Graham M. Snyder, MD, a fellow at Beth Israel Deaconess Hospital in Boston, Massachusetts. Erika D'Agata, MD, and Lara Venkataraman, PhD, also from Beth Israel Deaconess Hospital, were coauthors.
"Future research should be aimed at more accurate methods of surveillance than the swab used in our study and investigate whether there are groups of patients for whom the swab may be more accurate," Dr. Snyder added.
This study used standard agar plates infiltrated with ciprofloxacin or ceftazidime to screen for MDRGN bacteria. Other methods include PCR-based tests for carbapenemase-producing organisms and CHROMagar, he said. Perianal swabs were used because they are easy to perform and more comfortable than full rectal swabs, Dr. Snyder said. In addition to the rectum, potential sites for swabs include the groin, stool, wound, and perineum.
Thirty-five patients agreed to participate in the study, 3 of whom harbored 2 different species of MDRNG bacteria, for a total of 38 distinct MDRNG-patient pairs. The surveillance swab correctly identified 30 of the 38 (78.9%) MDRGN bacteria from clinical isolates. The perianal skin swab identified 1 or both MDRNG clinical isolates in 29 (82.9%) of the 35 patients.
Dr. Snyder gave some potential explanations for the findings. "Failure to detect 20% of MDRNG bacteria could be related to a time lag between identification of the organism initially and using a perianal swab. Or perhaps other sites were colonized and we didn't use a swab on those sites. It is also possible that our methods [microbiological plates] may not have been sensitive enough," he suggested.
"In the big picture, we need to carefully consider the methods we use for active surveillance. We need to think about the site of the swab and the methods we use," Dr. Snyder told Medscape Medical News.
More Optimistic View of Study
"I was impressed by the sensitivity of Dr. Snyder's method, which was around 80% in this study. The study consisted of patients already on antibiotics, which would theoretically have decreased the sensitivity of surveillance swabs to detect the organisms," said Mary-Claire Roghmann, MD, from the University of Maryland in Baltimore.
Dr. Roghmann told Medscape Medical News that she thought that the method of perianal swab testing could have higher sensitivity if performed prior to treatment with antibiotics, as occurred in this study.
"Based on this study, I actually think that the use of perianal swabs is promising," she stated.
Dr. Snyder and Dr. Roghmann have disclosed no relevant financial relationships.
50th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC): Abstract K-317. Presented September 12, 2010.
NDM-1 Gene Spreading to Multiple Bacteria Species, Making Them Antibiotic-Resistant
Alice Goodman
September 22, 2010 (Boston, Massachusetts) — The gene that encodes for New Delhi metallo-beta-lactamase-1 (NDM-1), which confers resistance to most currently available antibiotics, appears to be spreading from the Indian subcontinent (India, Pakistan, and Bangladesh), researchers reported here at a media press conference during the 50th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC).
NDM-1 has recently been documented in strains of bacteria in Australia, France, Japan, Kenya, North America, Singapore, Taiwan, and the United Kingdom. The emergence of this gene poses the threat of a pandemic with few treatment choices, said researchers.
In 2 separate reports at ICAAC, investigators described cases of NDM-1 in Escherichia coli in Canada and in Klebsiella sp. in Australia, prompting the news conference. Both case reports involved patients who had recently traveled to India. No one really knows the true prevalence of NDM-1-infiltrated bacteria, the researchers said, but they believe that the spread from India to other parts of the world is at least partly due to a confluence of a large Indian diaspora returning to their homeland for visits and to the phenomenon of medical tourism to India.
"The reservoir is in India, Pakistan, and Bangladesh, and is due to factors that are not controllable — overuse of antibiotics, poor hygiene, and diarrhea in an overcrowded overpopulated country. A plague could spread around the world, first through the Indian diaspora, which constitutes 20 million people all over the world," warned Patrice Nordmann, MD, from the Hôpital de Bicêtre, Le Kremlin-Bicêtre, France. "We feel it's only a question of time." Dr. Nordmann was a panelist at the press conference.
It is not known how widespread NDM-1 is in India, "which is one of our greatest concerns," added Timothy Walsh, MD, from Cardiff University in the United Kingdom.
"Our data are not accurate, but there are billions of people in India without clean water and sanitation. Massive antibiotic usage and antibiotics in the sewage create a nightmare that is a perfect recipe. Selective pressure fuels antibiotic resistance, and bugs are put on hyperdrive to accept DNA with NDM-1 encoded in plasmids," Dr. Walsh said.
Since the abstracts describing the 2 case reports of NMD-1-associated resistance were submitted to the ICAAC, other cases have emerged in Australia and Canada. Culture of the infection in Canada revealed the same strains of NMD-1 E coli identified in the United Kingdom and India. In the Australian case, NDM-1 was present in Klebsiella pneumoniae isolated from a foot wound.
"Both patients in Canada were treated successfully, but according to lab tests, only 3 antibioticswere found to have any effect — tigecycline, colistin, and phosphomycin," said senior author of that abstract, Johann Pitout, MD, from the University of Calgary in Alberta.
Perhaps more important is the fact that general practitioners are not used to seeing multidrug resistance associated with NDM-1, and if this type of infection does become common, infected patients may not get appropriate treatment, Dr. Pitout stated. "General practitioners are not qualified to treat these infections," he asserted.
September 22, 2010 (Boston, Massachusetts) — The gene that encodes for New Delhi metallo-beta-lactamase-1 (NDM-1), which confers resistance to most currently available antibiotics, appears to be spreading from the Indian subcontinent (India, Pakistan, and Bangladesh), researchers reported here at a media press conference during the 50th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC).
NDM-1 has recently been documented in strains of bacteria in Australia, France, Japan, Kenya, North America, Singapore, Taiwan, and the United Kingdom. The emergence of this gene poses the threat of a pandemic with few treatment choices, said researchers.
In 2 separate reports at ICAAC, investigators described cases of NDM-1 in Escherichia coli in Canada and in Klebsiella sp. in Australia, prompting the news conference. Both case reports involved patients who had recently traveled to India. No one really knows the true prevalence of NDM-1-infiltrated bacteria, the researchers said, but they believe that the spread from India to other parts of the world is at least partly due to a confluence of a large Indian diaspora returning to their homeland for visits and to the phenomenon of medical tourism to India.
"The reservoir is in India, Pakistan, and Bangladesh, and is due to factors that are not controllable — overuse of antibiotics, poor hygiene, and diarrhea in an overcrowded overpopulated country. A plague could spread around the world, first through the Indian diaspora, which constitutes 20 million people all over the world," warned Patrice Nordmann, MD, from the Hôpital de Bicêtre, Le Kremlin-Bicêtre, France. "We feel it's only a question of time." Dr. Nordmann was a panelist at the press conference.
It is not known how widespread NDM-1 is in India, "which is one of our greatest concerns," added Timothy Walsh, MD, from Cardiff University in the United Kingdom.
"Our data are not accurate, but there are billions of people in India without clean water and sanitation. Massive antibiotic usage and antibiotics in the sewage create a nightmare that is a perfect recipe. Selective pressure fuels antibiotic resistance, and bugs are put on hyperdrive to accept DNA with NDM-1 encoded in plasmids," Dr. Walsh said.
Since the abstracts describing the 2 case reports of NMD-1-associated resistance were submitted to the ICAAC, other cases have emerged in Australia and Canada. Culture of the infection in Canada revealed the same strains of NMD-1 E coli identified in the United Kingdom and India. In the Australian case, NDM-1 was present in Klebsiella pneumoniae isolated from a foot wound.
"Both patients in Canada were treated successfully, but according to lab tests, only 3 antibioticswere found to have any effect — tigecycline, colistin, and phosphomycin," said senior author of that abstract, Johann Pitout, MD, from the University of Calgary in Alberta.
Perhaps more important is the fact that general practitioners are not used to seeing multidrug resistance associated with NDM-1, and if this type of infection does become common, infected patients may not get appropriate treatment, Dr. Pitout stated. "General practitioners are not qualified to treat these infections," he asserted.
Antibiotic Prescribing Habits Have Shifted According to Pathogen Prevalence Patterns
Alice Goodman
September 22, 2010 (Boston, Massachusetts) — The choice of which antibiotic to prescribe as initial empiric therapy for hospitalized patients with complicated skin and skin-structure infections has changed substantially over the past decade, suggesting that physicians are responding to an increase in methicillin-resistant Staphylococcus aureus (MRSA), Pseudomonas aeruginosa, and Gram-negative infections in general. Multidrug use is also much more common than it was 10 years ago, according to a retrospective study reported here at the 50th Interscience Conference on Antimicrobial Agents and Chemotherapy.
The use of vancomycin — the most widely used drug for MRSA — increased dramatically, whereas the use of cefazolin and ampicillin/sulbactam — 2 drugs used to treat methicillin-susceptible S aureus — declined over the same period. Concomitantly, a marked increase was seen in prescribing pipericillin/tazobactam, a broad-spectrum antibiotic covering Gram-negative infections.
"These changes in initial antibiotic therapy in hospitalized patients undoubtedly reflect increasing concern about pathogen resistance and changes in treatment guidelines. Our objective was to see whether physicians are responding to the changing environment in infectious disease, and our study suggests that they are," said senior author David Weber, MD, professor of medicine, pediatrics, and epidemiology at the University of North Carolina in Chapel Hill.
The study used the Cerner Health Facts electronic database to identify 22,832 adult patients hospitalized for complicated skin and skin-structure infections in the United States between 2000 and 2009; 75% were hospitalized for acute infections, 18.3% for infections at the surgical site, and 6.6% for chronic infections.
A marked decrease over time was observed in the use of cefazolin (from 22.1% to 6.9%) and ampicillin/sulbactam (from 19.9% to 6.0%), whereas the use of vancomycin increased 4-fold (from 5.6% to 22.6%), and the use of piperacillin/tazobactam plus vancomycin increased more than 10-fold (from 0.9% to 12.9%. Piperacillin/tazobactam monotherapy increased more than 2-fold (from 2.8% to 6.5%).
Dr. Weber pointed out that the use of vancomycin as either a single agent or as part of a multidrug regimen increased from 16.5% in 2000 to 55.4% in 2009. During that time period, the use of piperacillin/tazobactam either as a single agent or as part of a multidrug regimen increased from 6.8% to 20.9%.
Over the same time period, the use of initial regimens providing coverage for P aeruginosa increased from 16.4% to 28.1%, and antibiotic regimens providing broad-spectrum coverage for Gram-negative infections increased from 22.1% to 36.1%.
Dr. Weber cited several limitations of this study: its retrospective design, the fact that its database might not be generalizable to all American hospitals, and the fact that it provided no information on why initial antibiotics were selected.
Commenting on this study, Vance Fowler, MD, associate professor at Duke University in Durham, North Carolina, said: "Multidrug-resistant pathogens are on the rise in critically ill patients who present to the hospital with life-threatening complications of skin and skin-structure infections. We want to make sure we are choosing antibiotics wisely."
"Physicians have to make an educated guess during the first 24 hours of hospitalization, and they get points for being right. You know antimicrobial resistance is climbing in the hospital setting, and your job is to ensure that the patient in the bed right now has the best chance to get the best therapy out of the gate," he continued.
"This study implies physicians are doing their job," Dr. Fowler stated.
Dr. Weber reports receiving financial support from Policy Analysis Inc and the Forest Research Institute of New York City. Dr. Fowler reports financial ties with Astellas, Cubist, Merck, Theravance, Pfizer, Novartis, Inhibitex, Arpida, Targanta, Wyeth, Ortho-McNeil, Vertex Pharmaceuticals, Leo Pharmaceuticals, NovaDigm, The Medicines Company, Baxter Pharmaceuticals, Biosynexus, and Johnson & Johnson.
50th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC): Abstract L1-1761. Presented September 14, 2010.
September 22, 2010 (Boston, Massachusetts) — The choice of which antibiotic to prescribe as initial empiric therapy for hospitalized patients with complicated skin and skin-structure infections has changed substantially over the past decade, suggesting that physicians are responding to an increase in methicillin-resistant Staphylococcus aureus (MRSA), Pseudomonas aeruginosa, and Gram-negative infections in general. Multidrug use is also much more common than it was 10 years ago, according to a retrospective study reported here at the 50th Interscience Conference on Antimicrobial Agents and Chemotherapy.
The use of vancomycin — the most widely used drug for MRSA — increased dramatically, whereas the use of cefazolin and ampicillin/sulbactam — 2 drugs used to treat methicillin-susceptible S aureus — declined over the same period. Concomitantly, a marked increase was seen in prescribing pipericillin/tazobactam, a broad-spectrum antibiotic covering Gram-negative infections.
"These changes in initial antibiotic therapy in hospitalized patients undoubtedly reflect increasing concern about pathogen resistance and changes in treatment guidelines. Our objective was to see whether physicians are responding to the changing environment in infectious disease, and our study suggests that they are," said senior author David Weber, MD, professor of medicine, pediatrics, and epidemiology at the University of North Carolina in Chapel Hill.
The study used the Cerner Health Facts electronic database to identify 22,832 adult patients hospitalized for complicated skin and skin-structure infections in the United States between 2000 and 2009; 75% were hospitalized for acute infections, 18.3% for infections at the surgical site, and 6.6% for chronic infections.
A marked decrease over time was observed in the use of cefazolin (from 22.1% to 6.9%) and ampicillin/sulbactam (from 19.9% to 6.0%), whereas the use of vancomycin increased 4-fold (from 5.6% to 22.6%), and the use of piperacillin/tazobactam plus vancomycin increased more than 10-fold (from 0.9% to 12.9%. Piperacillin/tazobactam monotherapy increased more than 2-fold (from 2.8% to 6.5%).
Dr. Weber pointed out that the use of vancomycin as either a single agent or as part of a multidrug regimen increased from 16.5% in 2000 to 55.4% in 2009. During that time period, the use of piperacillin/tazobactam either as a single agent or as part of a multidrug regimen increased from 6.8% to 20.9%.
Over the same time period, the use of initial regimens providing coverage for P aeruginosa increased from 16.4% to 28.1%, and antibiotic regimens providing broad-spectrum coverage for Gram-negative infections increased from 22.1% to 36.1%.
Dr. Weber cited several limitations of this study: its retrospective design, the fact that its database might not be generalizable to all American hospitals, and the fact that it provided no information on why initial antibiotics were selected.
Commenting on this study, Vance Fowler, MD, associate professor at Duke University in Durham, North Carolina, said: "Multidrug-resistant pathogens are on the rise in critically ill patients who present to the hospital with life-threatening complications of skin and skin-structure infections. We want to make sure we are choosing antibiotics wisely."
"Physicians have to make an educated guess during the first 24 hours of hospitalization, and they get points for being right. You know antimicrobial resistance is climbing in the hospital setting, and your job is to ensure that the patient in the bed right now has the best chance to get the best therapy out of the gate," he continued.
"This study implies physicians are doing their job," Dr. Fowler stated.
Dr. Weber reports receiving financial support from Policy Analysis Inc and the Forest Research Institute of New York City. Dr. Fowler reports financial ties with Astellas, Cubist, Merck, Theravance, Pfizer, Novartis, Inhibitex, Arpida, Targanta, Wyeth, Ortho-McNeil, Vertex Pharmaceuticals, Leo Pharmaceuticals, NovaDigm, The Medicines Company, Baxter Pharmaceuticals, Biosynexus, and Johnson & Johnson.
50th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC): Abstract L1-1761. Presented September 14, 2010.
jueves, 12 de agosto de 2010
BIOSÍNTESIS DEL PEPTIDOGLICANO

www.ugr.es/~eianez/Microbiologia/05paredbios.htm
1 INTRODUCCIÓN
En el capítulo anterior estudiamos la composición química, estructura y funciones de los principales tipos de paredes celulares procarióticas. En este capítulo trataremos un aspecto dinámico del tema de las paredes: cómo se sintetiza el peptidoglucano de las eubacterias, y cómo se produce el proceso general de crecimiento de la pared, incluyendo el evento particular de formación del septo transversal que marca el “nacimiento” de dos células hijas. Nos concentraremos en la biosíntesis del peptidoglucano, debido a su interés intrínseco y aplicado (sobre este proceso actúan diversos antibióticos, algunos de gran importancia clínica).
Desde el punto de vista topológico, todas las capas de las envueltas bacterianas (membranas, pared celular) son superficies cerradas sobre sí mismas, físicamente continuas para mantener la integridad y viabilidad de la célula.
Pero por otro lado, deben de ser susceptibles de expandirse durante el crecimiento por incorporación de nuevos materiales. Además, todos los constituyentes deben crecer coordinadamente e incorporarse en los lugares precisos. Por ejemplo, en el caso de las bacterias Gram-negativas, distintos componentes deben de ir a parar a diferentes localizaciones: periplasma, PG, membrana externa, e incluso medio exterior.
Durante cada ciclo celular, hay una fase en la que los materiales de las envueltas (y concretamente, de la pared celular que nos ocupa ahora) deben de facilitar la división de la célula en una descendencia de dos células hijas.
Finalmente, queda el problema de la energía. Los procesos biosintéticos requieren aporte de energía química, pero el ATP y compuestos similares no pueden salir del protoplasto.
Precisamente en este capítulo vamos a ver algunas de las estrategias bioquímicas y moleculares que las bacterias han evolucionado para solventar estos puntos para el caso del peptidoglucano. Veremos que la estrategia implica varias fases:
*Síntesis de precursores en el citoplasma
*Ensamblaje parcial en membrana
*Transporte a la cara externa externa de la membrana
*Ensamblaje final en el exterior, mediante reacciones que no precisan energía
2 BIOSÍNTESIS DEL PEPTIDOGLUCANO DE MUREÍNA
Consta de 4 etapas:
1.Síntesis de precursores solubles en el citoplasma.
2.Estos precursores son transferidos a un transportador lipídico situado en la membrana citoplásmica (un poliisoprenol fosfatado llamado undecaprenil-fosfato), donde se forman las unidades disacarídicas con el pentapéptido.
3.Las unidades disacarídicas se polimerizan en cadenas lineales fuera de la membrana, pero aún unidas al undecaprenil-fosfato de la membrana.
4.Unión del polímero lineal así formado al peptidoglucano preexistente en la pared celular, por entrecruzamiento de (al menos) parte de sus péptidos respectivos.
A estas etapas hay que añadir una fase adicional de regeneración del transportador lipídico, una vez que ha cumplido su misión, para que pueda ser operativo en un nuevo ciclo de síntesis.
Veamos, pues, en más detalle, cómo ocurre este interesante proceso:
Fase 1:. Los monosacáridos que luego van a constituir la unidad disacarídica repetitiva del esqueleto del peptidoglucano (NAM y NAG) se activan al unirse a uridín difosfato (UDP). (En general, los monosacáridos que han de incorporarse a polímeros de pared celular bacteriana se activan mediante su unión con nucleósidos-fosfato.)
Por lo tanto, en esta fase se sintetizan por separado:
NAG-UDP
NAM-UDP
Luego se va produciendo la adición secuencial y ordenada de los distintos aminoácidos al NAM (en reacciones que requieren energía e iones Mn++):
1. L-ala
2. D-glu
3. m-DAP (u otro diaminoácido; p. ej. L-lys en Staphylococcus aureus)
4. D-ala-D-ala
Observar que no se produce un tetrapéptido, sino un pentapétido. El último paso de adición de aminoácidos es la unión del dipéptido D-alanil-D-alanina, que se ha sintetizado en dos fases:
una racemasa convierte la L-ala a D-ala;
creación de enlace peptídico entre dos D-ala.
Fase 2: El UDP-NAM-pentapéptido se transfiere ahora a un transportador de membrana, llamado undecaprenil-fosfato (que abreviaremos como Lip-P), en una reacción catalizada por una translocasa específica.
El undecaprenil-fosfato es un poliisoprenoide de 55 átomos de C (C55, derivado de la repetición 11 veces de la unidad isoprenoide, con un fosfato terminal). Se le conoce también con el nombre de bactoprenol, pero hoy se sabe que no es exclusivo de bacterias. El bactoprenol permite el transporte y ensamblaje de sustancias que, como los azúcares, son hidrofílicas, y no podrían pasar por sí mismas la barrera hidrofóbica de la membrana.
Una vez que el NAM-pentapétido está unido al undecaprenil (por medio de pirofosfato), una transferasa transfiere a éste la NAG desde el UDP-NAG. Se genera pues el enlace ß(1à4) entre NAG y NAM. Por lo tanto, se obtiene: Lip-P-P-NAM(pentapéptido)-NAG.
En esta situación es cuando se producen la modificaciones que ya estudiamos en la estructura básica del PG. Por ejemplo: en Staphylococcus aureus el grupo -COOH del D-glutámico en posición (2) es amidado (pasa a --CO-NH2. Por otro lado, se introducen los puentes peptídicos, que en el caso de esta bacteria consisten en una pentaglicina, que se une al grupo amino terminal de la L-Lys en posición (3).
Tanto la traslocasa como la transferasa está localizadas en el lado citoplásmico de la membrana, de modo que el precursor Lip-P-P-NAM(pentapéptido)-NAG, en este momento está “colgando” hacia el citoplasma, anclado a la lámina interna de la membrana a través de bactoprenol.
Fase 3: Polimerización de varias unidades disacarídicas: Ahora el bactoprenol “se da la vuelta” en la membrana (una especie de flip-flop desde la capa interna hasta la externa), de modo que logra que el precursor resultante de la fase 2 quede expuesto hacia el medio acuoso exterior a la membrana. Entonces tiene lugar la polimerización de varias unidades disacarídicas: ello se logra en una reacción de transglucosidación. Consiste en la unión de cada unidad disacarídica (con su pentapéptido) unida a su respectivo Lip-P-P, con el extremo libre (reductor) de una cadena preexistente que a su vez está unida a otra molécula de Lip-P-P.
En el proceso se libera uno de los Lip-P-P (o sea, el undecaprenil, pero en forma pirofosforilada). Sobre este Lip-P-P actúa una fosfatasa específica, que elimina el fosfato terminal, regenerándose el undecaprenil-fosfato, que queda dispuesto para otro ciclo como el descrito.
Fase 4: El polímero surgido de la fase anterior es una cadena lineal de PG sin entrecruzar, y unido aún al transportador lipídico de membrana. Ahora este polímero naciente (con sus pentapéptidos) reacciona, por transpeptidación, con un PG aceptor preexistente. En esta reacción se ven implicados el grupo C=O de la D-ala (4) del PG naciente y el grupo -NH2 libre del diaminoácido (3) del PG aceptor (o del último aminoácido del puente peptídico).
Esto es lo mismo que decir que el enlace peptídico entre D-ala (4) y D-ala (5) del PG naciente se ve sustituido por otro enlace peptídico, entre dicha D-ala (4) y el diaminoácido del PG naciente.
La energía para esta reacción la suministra la hidrólisis concomitante del enlace peptídico entre las dos D-ala terminales. Es decir, en cada reacción de transpeptidación se libera una D-ala, correspondiente a la que ocupaba la posición (5).
Ya dijimos en el capítulo anterior que no todos los tetrapéptidos participan en entrecruzamientos. Las D-ala terminales (en 5) de los péptidos no implicados en tales entrecruzamientos son eliminadas por una enzima llamada D-D-carboxipeptidasa. Esta enzima explica no sólo que en el PG maduro existan tetrapéptidos (y no los pentapéptidos originales), sino también la existencia de tripéptidos.
Muchas bacterias controlan el grado de entrecruzamiento de su PG maduro. Incluso algunas pueden eliminar totalmente muchos de los péptidos originalmente unidos al NAM, mediante enzimas conocidas genéricamente con el nombre de autolisinas. Así por ejemplo, Micrococcus luteus -un coco Gram-positivo- merced a su actividad NAM-L-ala-amidasa, presenta un PG que no está entrecruzado en un 50-70%.
Antibióticos que actúan a nivel de la biosíntesis de peptidoglucano:
Estos antibióticos tienen un efecto bactericida sobre bacterias en crecimiento. Ello se debe a que, al inhibir determinados pasos del ciclo de síntesis y ensamblaje del PG, provocan la acumulación de precursores de dicho PG, lo que a su vez desencadena la activación de las autolisinas de la bacteria, que degradan el PG y que finalmente provoca la lisis celular (en medios hipotónicos), por entrada masiva de agua a la célula.
1.Fosfomicina: actúa inhibiendo la formación del 3-O-D-lactil-éter de la NAG (o sea, del NAM). Parece ser que la base molecular estriba en la semejanza estructural entre el este antibiótico y el PEP (es decir, la fosfomicina es análogo estructural del PEP, lo que lleva a la inactivación de la enzima correspondiente a esta reacción).
2.Cicloserina: Se comporta como análogo estructural de la D-alanina, por lo que inhibe la actuación de la racemasa que convierte la L-ala a D-ala, así como de la reacción de unión de dos D-ala.
3.Tunicamicina: inhibe la traslocasa que cede el NAM unido hasta entonces al UDP y lo pasa al bactoprenol (fase 2ª).
4.Vancomicina y ristocetina: inhiben la segunda transglucosidación (fase 3ª), es decir, la unión de diversas unidades disacarídicas.
5.Bacitracina: se une al undecaprenol-pirofosfato, bloqueando su desfosforilación, e impidiendo por lo tanto, la regeneración del transportador de membrana.
6.Antibióticos ß-lactámicos (p. ej.: penicilinas, cefalosporinas): inhiben la reacción de entrecruzamiento por transpeptidación. (Estudiaremos su mecanismo de acción en más detalle en el capítulo 20 sobre Quimioterápicos y Antibióticos).
3 CRECIMIENTO DE LA PARED CELULAR
Como ya dijimos al comienzo de este tema, aunque la pared celular es una estructura cerrada y sin solución de continuidad, debe permitir su expansión (crecimiento), y esto supone que se han de romper ciertos enlaces si se quiere que el nuevo material se ensamble con el preexistente mediante nuevas uniones, es decir, debe existir una acción concertada de mureín-hidrolasas y de mureín-sintasas. Por otro lado, hay que tener en cuenta que en el crecimiento de la pared celular se producen dos tipos de procesos: la expansión (aumento de tamaño) de esa pared, y la formación del tabique transversal en el centro de la célula.
El crecimiento y septación del peptidoglucano están basados en la actividad controlada y localizada en puntos determinados, de una gama de autolisinas, especialmente las dotadas de actividad transglucosidasa y/o transpeptidasa. Debido a que estas enzimas son los sitios de acción de las penicilinas (y otros antibióticos ß-lactámicos), se les conoce también con el nombre de PBP (de penicillin-binding proteins; véase la tabla 1).
3.1 CRECIMIENTO DE LA PARED EN UNA BACTERIA GRAM-NEGATIVA: Escherichia coli
El crecimiento de la pared en E. coli se puede considerar dividido en dos fases:
1. Crecimiento en longitud (elongación), mientras la célula crece en tamaño.
2. Producción del tabique transversal (septación), que conduce a la formación de dos células hijas.
Elongación
Las PBPs 1 son las encargadas de la elongación de las cadenas de PG naciente (por transglucosidación de las unidades disacarídicas) y simultáneamente, por transpeptidación, logran el entrecruzamiento.
Las cadenas nacientes del PG se intercalan en la parte cilíndrica del sáculo de PG gracias a que las PBP4 y PBP5 cortan enlaces del PG preexistente. La PBP2 interviene aquí también para transpeptidación. La cadena de PG se va elongando unidireccionalmente alrededor de la circunferencia de la célula. Se calcula que existen unos 200 sitios de inserción de nuevo material en cada célula, dispersos de forma más o menos uniforme por toda la superficie de la célula. La maquinaria biosintética tarda 8 minutos en completar cada circunferencia de elongación.
Formación del tabique transversal
La división de la bacteria por fisión binaria simétrica se logra por medio de una invaginación circular de las envueltas (membrana citoplásmica y peptidoglucano) en mitad de la célula madre. En el inicio de este proceso tiene un papel esencial la proteína FtsZ, y más tarde intervienen otras proteínas Fts.
FtsZ es una proteína imprescindible, presente tanto en eubacterias como en arqueas, que muestra parecido con las tubulinas eucarióticas. Al parecer, al igual que las tubulinas, la FtsZ se une a GTP, con lo que se promueve su polimerización. Bajo control del ciclo celular, la FtsZ se ensambla poco antes de la división en el centro de la célula, formando un “anillo citocinético” en el lado citoplásmico de la membrana celular. Existen indicios fuertes de que la formación de este anillo de FtsZ señaliza el sitio por donde se producirá la división y se activará el crecimiento del peptidoglucano del tabique transversal. Una vez que FtsZ ocupa el lugar del futuro septo, entran en acción otras proteínas Fts, formando un complejo llamado “divisoma”.
En las fotografías a microscopio electrónico se ve cómo bajo la invaginación de membrana que constituye la “avanzadilla” del septo, se localizan las moléculas de FtsZ.
La hipótesis señala que los polímeros de FtsZ se van “contrayendo”, de modo que “tiran” de las envueltas hacia el interior, provocando la típica invaginación alrededor del centro del bacilo, y que simultáneamente, la FtsZ provoca la activación de la PBP3 (=FtsI), que es una transglucosidasa/transpeptidasa específica del tabique transversal.
A microscopio electrónico se observa que este tabique está formado por dos láminas de PG (densas a los electrones) separadas entre sí por otra transparente. El final de la división ocurre como consecuencia de la invaginación de la membrana externa entre las dos láminas de PG.
¿Cómo se ensambla la membrana externa con relación al PG? Parece ser que esta membrana se ensambla en buena medida por procesos espontáneos guiados por interacciones entre el LPS y proteínas, sirviendo el PG subyacente como andamio que facilita el montaje de toda la estructura. Parece ser que las zonas de adhesión (junturas de Bayer) son importantes para la correcta colocación de LPS y proteínas de la membrana externa.
3.2 CRECIMIENTO Y SEPTACIÓN EN UNA BACTERIA GRAM-POSITIVA: Enterococcus faecalis
A diferencia de lo visto en E. coli, en el enterococo (Enterococcus faecalis) el crecimiento del PG no es difuso, sino zonal, comenzando en el centro (zona ecuatorial) y avanzando hacia afuera.
Véase en la figura el experimento de marcado de pared celular con anticuerpos fluorescentes, con ulterior seguimiento del marcaje al microscopio:
tras unos 15 minutos después del marcado se observa que existe una banda ecuatorial oscura (no marcada, por lo tanto está constituida por material nuevo recién insertado), mientras que los polos de las células siguen enteramente marcados.
Tras 30 o 60 minutos observamos células enteramente sin marcar, intercaladas entre células con uno de sus polos marcados.
El proceso se puede describir así:
1.En la célula adulta antes de la división aparece una banda ecuatorial donde la pared celular se hace más gruesa.
2.Debajo de esta zona engrosada (hacia el interior del citoplasma) se va depositando nuevo material, lo que marca el inicio de la formación del tabique transversal. Enseguida aparece una muesca en la banda ecuatorial.
3.El tabique, con un grosor doble al de la pared celular de la superficie celular, sigue avanzando, hasta que...
4.Se completa. Simultáneamente a los pasos 3º y 4º la zona de nueva síntesis de P.C. superficial alcanza el ecuador de las celúlas hijas nacientes.
5.El tabique completo de doble grosor va siendo escindido en dos mitades desde el exterior hacia el centro, por acción de autolisinas. De esta forma se le adjudica a cada célula hija la nueva pared correspondiente a uno de sus polos (el otro procede, intacto, de la célula madre).
Véase igualmente la figura que muestra en más detalle la formación del tabique. La idea que se saca del proceso en esta bacteria es que posee una sola zona de crecimiento de PG, con dos regiones de deposición de nuevo material: la región del tabique transversal propiamente dicho, y la región adyacente, ya en la superficie celular, responsable de la expansión de la pared periférica, a ambos lados del tabique.
4 PROTOPLASTOS Y ESFEROPLASTOS
Mediante procedimientos de laboratario se puede lograr eliminar total o parcialmente la pared celular bacteriana. Se denominan protoplastos las células bacterianas a las que se ha desprovisto totalmente de pared celular, mientras que esferoplastos son aquellas células bacterianas que poseen restos de pared.
Obtención. Existen dos posibles métodos alternativos:
1.Por destrucción del entramado del PG mediante enzimas líticas (lisozima, peptidasas). En el caso de bacterias Gram-negativas, previamente hay que desorganizar la membrana externa para hacerla permeable a estas enzimas. Ello se logra usando el quelante EDTA y/o sometiendo las células a bajas temperaturas.
2.Por inhibición de la formación de nuevo PG en las células en crecimiento, tratándolas p. ej., con penicilina. Si se parte de un mutante auxotrofo para un componente del PG basta hacer crecer a la bacteria en un medio carente de dicho componente.
Estos métodos permiten:
en el caso de Gram-positivas, la desorganización total de su pared, por lo que se obtienen protoplastos;en el caso de las Gram-negativas, quedan restos de membrana externa y de peptidoglucano atrapados en ella, por lo que se obtienen esferoplastos.
En ambos casos, protoplastos y esferoplastos pueden revertir a la forma normal eliminando el tratamiento (retirando la lisozima, o la penicilina).
Protoplastos y esferoplastos son formas osmóticamente sensibles debido precisamente a que al no existir o estar desorganizado parcialmente el PG, ya no se pueden contrarrestar las fuerzas de presión osmótica existentes en los medios hipotónicos en que normalmente se cultivan o suspenden las bacterias. Por lo tanto, si se quieren obtener suspensiones estables de estas formas, hay que obtenerlas en medios o soluciones isotónicos o ligeramente hipertónicos, para evitar su lisis:
soluciones de NaCl 0,25-0,5 M;
sorbitol o sacarosa 0,1-0,5 M;
polietilénglicol (PEG) al 7,5%.
En estos medios los protoplastos y esferoplastos poseen formas esféricas, independientemente de la forma que tuviera la bacteria con pared de que proceden.
Se emplean en varios aspectos de investigación básica:
método suave para luego obtener extractos libres de células y fracciones subcelulares.
en algunas bacterias, método para hacerlas permeables al ADN, en experimentos de transformación genética.
para realizar fusión entre protoplastos de diferentes cepas de la misma especie e incluso entre especies distintas. Se trata de un método de obtención de “recombinantes somáticos” usado para determinados estudios genéticos en bacterias que no tengan sistemas naturales de transferencia genética.
5 FORMAS L
Son células bacterianas carentes total o casi totalmente de P.C., con formas pleomórficas, irregulares y globulares, que se producen de forma espontánea en algunas especies bacterianas (p. ej., Streptobacillus moniliformis) cuando se cultivan en medios a base de suero (que son hipertónicos).
Las colonias de las formas L naturales son muy características: en “huevo frito”, bifásicas.
Se pueden obtener de forma inducida formas L en diversas bacterias Gram-positivas y Gram-negativas, tratándolas con penicilina en medios hipertónicos.
Las formas L inestables se generan por tratamientos breves, y pueden revertir con frecuencia a la forma normal con pared. Poseen algo de PG, aunque éste se encuentra alterado respecto al PG de la célula normal.
Las formas L estables se producen por tratamientos más prolongados, y no suelen revertir al tipo normal. La mayoría carecen totalmente de peptidoglucano.
Así pues, en las formas L el peptidoglucano ha sido eliminado total o parcialmente, pero de manera que ya no puede servir de aceptor de nuevo PG.
BIBLIOGRAFÍACAPÍTULOS DE LIBROS DE REFERENCIA
VAZQUEZ, D. (1986): Biosíntesis del peptidoglucano: mecanismos de acción y selectividad de los antibióticos inhibidores. En: Bioquímica y Biología Molecular (Coordinador: L. Cornudella). Salvat, Barcelona, págs. 133-140.
WHITE, D. (1995): “The physiology and biochemistry of Prokaryotes”. Oxford University Press, Nueva York y Oxford. Consultar el capítulo 10.
ARTÍCULOS DE REVISIÓN
KENNEDY, E.P. (1996): Membrane-derived oligosaccharides. En: “Escherichia coli and Salmonella typhimurium. Cellular and molecular biology”, 2ª edición (F.C. Neidhart, ed.). American Society for Microbiology Press. Washington, D.C. (1996), págs. 1064-1071.
LUTKENHAUS, L. (1993): FtsZ ring in bacterial cytokinesis. Mol. Microbiol. 9: 403-409.
LUTKENHAUS, J., A. MUKHEEERJEE (1996): Cell division. En: “Escherichia coli and Salmonella typhimurium. Cellular and molecular biology”, 2ª edición (F.C. Neidhart, ed.). American Society for Microbiology Press. Washington, D.C. (1996), págs. 1615-1625.
NANNINGA, N. (1991): Cell division and peptidoglycan assembly in Escherichia coli. Mol. Microbiol. 5: 791.795.
NANNINGA, N., F.B. WIENTJES, E. MULDER, C.L. WOLDRINGH (1992): Envelope growth in Escherichia coli. Spatial and temporal organization. En "Prokaryotic structure and function: a new perspective. Society for General Microbiology & Cambridge University Press, Cambridge, pp. 185-221.
RAETZ, CH.R.H. (1996) Bacterial lipopolysaccharides: a remarkable family of bioactive macroamphiphiles. En: “Escherichia coli and Salmonella typhimurium. Cellular and molecular biology”, 2ª edición (F.C. Neidhart, ed.). American Society for Microbiology Press. Washington, D.C. (1996), págs. 1035-1063.
VAN HEIJENOORT, J. (1996): Murein synthesis. En: “Escherichia coli and Salmonella typhimurium. Cellular and molecular biology”, 2ª edición (F.C. Neidhart, ed.). American Society for Microbiology Press. Washington, D.C. (1996), págs. 1025-1034.
VICENTE, M., J. ERRINGTON (1996): Struture, function and controls in microbial division. Mol. Microbiol. 20: 1-7.
VINELLA, D., P. BOULOC, R. D'ARI (1993): GTPase enters the ring. Curr. Biol. 3: 65-66.
lunes, 5 de julio de 2010
Antibiotics May Not Be Needed After Abscess Incision and Drainage
News Author: Karla Gale, MS
CME Author: Penny Murata, MD
Authors and Disclosures
CME Released: 04/09/2010; Valid for credit through 04/09/2011
April 9, 2010 — Antibiotics don't improve outcomes after incision and drainage of uncomplicated skin abscesses, new research indicates. They might prevent new abscesses at one month, however.
"We're seeing so many abscesses now," lead researcher Dr. Gillian R. Schmitz told Reuters Health. Before methicillin-resistant Staphylococcus aureus (MRSA) became so widespread, most abscess patients had diabetes or were immunocompromised, she said, "but now they're popping up all over the place in people with no traditional risk factors."
"But no one has studied best practice for treating abscesses," Dr. Schmitz added.
In a multicenter trial, she and her colleagues randomized 212 adults to receive trimethoprim-sulfamethoxazole (160 mg/800 mg) or placebo after incision and drainage of community-acquired abscesses. They instructed patients to take 2 pills twice daily for seven days. The primary outcome was treatment failure at 7 days, defined as no improvement after 2 days, development of a new separate abscess within 7 days, or worsening infection within 7 days requiring intervention.
Eighty-eight patients in the antibiotic group and 102 in the placebo group completed 7 days of follow-up; 46 and 50, respectively, returned at 30 days.
According to their March 29th online report in the Annals of Emergency Medicine, all bacterial isolates were uniformly sensitive to trimethoprim-sulfamethoxazole. Still, there was no significant difference in treatment failure rates at 7 days (17% in the antibiotic group and 26% in the placebo group, p = 0.12).
"Antibiotics don't help with resolution of infection," Dr. Schmitz said. "The most important thing is to open the wound, clean it, and get the pus out."
However, the total number of new lesions at 30 days was significantly lower in the antibiotic group: 9% vs 28% (p = 0.02).
"More study needs to be done to draw a conclusion about recurrence, because we lost so many to follow-up at 30 days," Dr. Schmitz said.
A separate trial in children with community-acquired skin abscesses found similar results. In a paper scheduled for print publication in the May issue of same journal, Dr. Myto Duong, currently at Southern Illinois University, Springfield, and associates report outcomes in 149 children after incision and drainage. The 73 children randomized to the intervention group took trimethoprim-sulfamethoxazole for 10 days (10-12 mg trimethoprim/kg/day in 2 divided doses, with a maximum of 160 mg per dose, in a liquid formulation containing 200 mg sulfamethoxazole/40 mg trimethoprim per 5 mL). The other 76 children received placebo.
The authors found no significant difference in failure rates at 10 days — 4.1% in the antibiotic group vs 5.3% in the placebo group. In addition, the antibiotic group had a significantly lower incidence of new lesions at day 10 (12.9%, vs 26.4% in the placebo group).
At 30 days, however, with 46 children in the antibiotics group and 52 in the placebo group evaluable, there was no longer a significant difference in the rate of new lesions (28.3% vs 28.8%, respectively).
"Antibiotics are not required for pediatric skin abscess resolution," Dr. Duong's group concludes, adding that further research is needed to see if antibiotics help prevent new lesions in the short term.
Both studies were limited by attrition of patients and by inclusion of subjects who were otherwise healthy, so the results can't be generalized to patients with comorbidities. In Dr. Duong's trial, medication compliance was only 66%.
In an editorial, Dr. David A. Talan of Olive View--UCLA Medical Center in Sylmar, California, states, "Antibiotics for drained simple abscesses are not required to meet the standard of care."
He told Reuters Health, "The rate of resolution of patient groups described in these studies is very high, so doctors can feel that the odds are on their side" if they forego antibiotic treatment, "and they can reassure patients there's a good chance that things will work out fine."
But, he continued, physicians should be aware of the tendency for abscesses to return, and they should advise patients to seek attention if new symptoms appear after first infection resolves.
Ann Emerg Med. Published online March 29, 2010.
Reuters Health Information 2010. © 2010 Reuters Ltd.
Clinical Context
In the November 2007 issue of Antimicrobial Agents and Chemotherapy, Rajendran and colleagues reported that after incision and drainage for abscess, improvement at 7 days occurred in 84% of patients receiving antibiotics and 91% of patients receiving placebo. In children with skin abscesses, rates of treatment failure were similar for those treated with trimethoprim-sulfamethoxazole vs placebo, as found by Duong and colleagues in the May 5, 2009, online issue of the Annals of Emergency Medicine.
This multicenter, double-blind, randomized, placebo-controlled trial evaluates whether treatment with trimethoprim-sulfamethoxazole for uncomplicated skin abscesses reduces treatment failure in the first 7 days and the rate of new lesion formation in the first 30 days after incision and drainage in adults.
Study Highlights
• 212 adults 16 years or older from a convenience sample of 220 adults with uncomplicated skin abscesses requiring incision and drainage were enrolled.
• 4 military emergency departments participated.
• Exclusion criteria were immunocompromised status, pregnancy, breast-feeding, allergy to sulfa drugs, fever or systemic illness, antibiotic use in the prior week, hospitalization in the prior month, abscess on the face, suspected tracts or fistulas, and need for drainage in the operating room.
• Standard practice was used to drain and pack abscesses.
• Incision length, irrigation, and ultrasonography use were dependent on the individual physician.
• Wound cultures were sent for aerobic and anaerobic cultures and antimicrobial susceptibility testing.
• 96 patients were randomly assigned to receive trimethoprim-sulfamethoxazole (160 mg/800 mg) 2 pills twice daily for 7 days.
• 116 patients were randomly assigned to receive placebo.
• The placebo group vs the trimethoprim-sulfamethoxazole group had similar baseline characteristics of age (range, 17 - 79 years), sex, abscess location, cellulitis size, and abscess size.
• The most common abscess location was an extremity (47% - 49%).
• Patients were asked to follow up in the emergency department at 2 days and again at 7 days for wound check.
• Primary outcome measure was treatment failure within 7 days.
• Treatment failure in 7 days was defined as no improvement after 2 days, new separate lesion within 7 days, or worsening infection within 7 days requiring intervention (increased diameter of abscess, cellulitis, fever, or systemic response requiring further antibiotic use, additional incision and drainage, surgical debridement, or hospital admission).
• Secondary outcome measure was formation of new lesions, abscess, or pustule within 30 days.
• 3 sites assessed secondary outcome by telephone calls, return visits to the emergency department, and review of medical records.
• Culture results revealed MRSA in 53% of patients overall: 47 (47%) of 100 patients in the placebo group and 50 (60%) of 84 patients in the trimethoprim-sulfamethoxazole group.
• All MRSA isolates were sensitive to trimethoprim-sulfamethoxazole.
• At 7-day follow-up of 190 (90%) of 212 subjects, the treatment failure rate was statistically similar for the placebo group (27 [26%] of 102 patients) vs the trimethoprim-sulfamethoxazole group (15 [17%] of 88 patients). The difference was 9% (P = .12).
• At 30-day follow-up of 96 (69%) of 139 subjects, new lesion formation was greater in the placebo group (14 [28%] of 50 patients) vs the trimethoprim-sulfamethoxazole group (4 [9%] of 46 patients). The difference was 19% (P = .02).
• Adverse effects were reported by 10 patients in the trimethoprim-sulfamethoxazole group: 4 had nausea, 3 were drowsy or dizzy, 1 had headache and night sweats, 1 had vaginal yeast infection, and 1 had mild allergic reaction.
• 1 patient in the placebo group reported an adverse effect of drowsiness.
• Limitations of the study were loss to follow-up, convenience sample, inability to generalize to children or to persons with compromised immune systems, and lack of standardization of incision-and-drainage technique.
Clinical Implications
• In adults with uncomplicated skin abscesses, trimethoprim-sulfamethoxazole does not decrease treatment failure at 7 days after incision and drainage.
• In adults with uncomplicated skin abscesses, trimethoprim-sulfamethoxazole might decrease the formation of new lesions at 30 days after incision and drainage
CME Author: Penny Murata, MD
Authors and Disclosures
CME Released: 04/09/2010; Valid for credit through 04/09/2011
April 9, 2010 — Antibiotics don't improve outcomes after incision and drainage of uncomplicated skin abscesses, new research indicates. They might prevent new abscesses at one month, however.
"We're seeing so many abscesses now," lead researcher Dr. Gillian R. Schmitz told Reuters Health. Before methicillin-resistant Staphylococcus aureus (MRSA) became so widespread, most abscess patients had diabetes or were immunocompromised, she said, "but now they're popping up all over the place in people with no traditional risk factors."
"But no one has studied best practice for treating abscesses," Dr. Schmitz added.
In a multicenter trial, she and her colleagues randomized 212 adults to receive trimethoprim-sulfamethoxazole (160 mg/800 mg) or placebo after incision and drainage of community-acquired abscesses. They instructed patients to take 2 pills twice daily for seven days. The primary outcome was treatment failure at 7 days, defined as no improvement after 2 days, development of a new separate abscess within 7 days, or worsening infection within 7 days requiring intervention.
Eighty-eight patients in the antibiotic group and 102 in the placebo group completed 7 days of follow-up; 46 and 50, respectively, returned at 30 days.
According to their March 29th online report in the Annals of Emergency Medicine, all bacterial isolates were uniformly sensitive to trimethoprim-sulfamethoxazole. Still, there was no significant difference in treatment failure rates at 7 days (17% in the antibiotic group and 26% in the placebo group, p = 0.12).
"Antibiotics don't help with resolution of infection," Dr. Schmitz said. "The most important thing is to open the wound, clean it, and get the pus out."
However, the total number of new lesions at 30 days was significantly lower in the antibiotic group: 9% vs 28% (p = 0.02).
"More study needs to be done to draw a conclusion about recurrence, because we lost so many to follow-up at 30 days," Dr. Schmitz said.
A separate trial in children with community-acquired skin abscesses found similar results. In a paper scheduled for print publication in the May issue of same journal, Dr. Myto Duong, currently at Southern Illinois University, Springfield, and associates report outcomes in 149 children after incision and drainage. The 73 children randomized to the intervention group took trimethoprim-sulfamethoxazole for 10 days (10-12 mg trimethoprim/kg/day in 2 divided doses, with a maximum of 160 mg per dose, in a liquid formulation containing 200 mg sulfamethoxazole/40 mg trimethoprim per 5 mL). The other 76 children received placebo.
The authors found no significant difference in failure rates at 10 days — 4.1% in the antibiotic group vs 5.3% in the placebo group. In addition, the antibiotic group had a significantly lower incidence of new lesions at day 10 (12.9%, vs 26.4% in the placebo group).
At 30 days, however, with 46 children in the antibiotics group and 52 in the placebo group evaluable, there was no longer a significant difference in the rate of new lesions (28.3% vs 28.8%, respectively).
"Antibiotics are not required for pediatric skin abscess resolution," Dr. Duong's group concludes, adding that further research is needed to see if antibiotics help prevent new lesions in the short term.
Both studies were limited by attrition of patients and by inclusion of subjects who were otherwise healthy, so the results can't be generalized to patients with comorbidities. In Dr. Duong's trial, medication compliance was only 66%.
In an editorial, Dr. David A. Talan of Olive View--UCLA Medical Center in Sylmar, California, states, "Antibiotics for drained simple abscesses are not required to meet the standard of care."
He told Reuters Health, "The rate of resolution of patient groups described in these studies is very high, so doctors can feel that the odds are on their side" if they forego antibiotic treatment, "and they can reassure patients there's a good chance that things will work out fine."
But, he continued, physicians should be aware of the tendency for abscesses to return, and they should advise patients to seek attention if new symptoms appear after first infection resolves.
Ann Emerg Med. Published online March 29, 2010.
Reuters Health Information 2010. © 2010 Reuters Ltd.
Clinical Context
In the November 2007 issue of Antimicrobial Agents and Chemotherapy, Rajendran and colleagues reported that after incision and drainage for abscess, improvement at 7 days occurred in 84% of patients receiving antibiotics and 91% of patients receiving placebo. In children with skin abscesses, rates of treatment failure were similar for those treated with trimethoprim-sulfamethoxazole vs placebo, as found by Duong and colleagues in the May 5, 2009, online issue of the Annals of Emergency Medicine.
This multicenter, double-blind, randomized, placebo-controlled trial evaluates whether treatment with trimethoprim-sulfamethoxazole for uncomplicated skin abscesses reduces treatment failure in the first 7 days and the rate of new lesion formation in the first 30 days after incision and drainage in adults.
Study Highlights
• 212 adults 16 years or older from a convenience sample of 220 adults with uncomplicated skin abscesses requiring incision and drainage were enrolled.
• 4 military emergency departments participated.
• Exclusion criteria were immunocompromised status, pregnancy, breast-feeding, allergy to sulfa drugs, fever or systemic illness, antibiotic use in the prior week, hospitalization in the prior month, abscess on the face, suspected tracts or fistulas, and need for drainage in the operating room.
• Standard practice was used to drain and pack abscesses.
• Incision length, irrigation, and ultrasonography use were dependent on the individual physician.
• Wound cultures were sent for aerobic and anaerobic cultures and antimicrobial susceptibility testing.
• 96 patients were randomly assigned to receive trimethoprim-sulfamethoxazole (160 mg/800 mg) 2 pills twice daily for 7 days.
• 116 patients were randomly assigned to receive placebo.
• The placebo group vs the trimethoprim-sulfamethoxazole group had similar baseline characteristics of age (range, 17 - 79 years), sex, abscess location, cellulitis size, and abscess size.
• The most common abscess location was an extremity (47% - 49%).
• Patients were asked to follow up in the emergency department at 2 days and again at 7 days for wound check.
• Primary outcome measure was treatment failure within 7 days.
• Treatment failure in 7 days was defined as no improvement after 2 days, new separate lesion within 7 days, or worsening infection within 7 days requiring intervention (increased diameter of abscess, cellulitis, fever, or systemic response requiring further antibiotic use, additional incision and drainage, surgical debridement, or hospital admission).
• Secondary outcome measure was formation of new lesions, abscess, or pustule within 30 days.
• 3 sites assessed secondary outcome by telephone calls, return visits to the emergency department, and review of medical records.
• Culture results revealed MRSA in 53% of patients overall: 47 (47%) of 100 patients in the placebo group and 50 (60%) of 84 patients in the trimethoprim-sulfamethoxazole group.
• All MRSA isolates were sensitive to trimethoprim-sulfamethoxazole.
• At 7-day follow-up of 190 (90%) of 212 subjects, the treatment failure rate was statistically similar for the placebo group (27 [26%] of 102 patients) vs the trimethoprim-sulfamethoxazole group (15 [17%] of 88 patients). The difference was 9% (P = .12).
• At 30-day follow-up of 96 (69%) of 139 subjects, new lesion formation was greater in the placebo group (14 [28%] of 50 patients) vs the trimethoprim-sulfamethoxazole group (4 [9%] of 46 patients). The difference was 19% (P = .02).
• Adverse effects were reported by 10 patients in the trimethoprim-sulfamethoxazole group: 4 had nausea, 3 were drowsy or dizzy, 1 had headache and night sweats, 1 had vaginal yeast infection, and 1 had mild allergic reaction.
• 1 patient in the placebo group reported an adverse effect of drowsiness.
• Limitations of the study were loss to follow-up, convenience sample, inability to generalize to children or to persons with compromised immune systems, and lack of standardization of incision-and-drainage technique.
Clinical Implications
• In adults with uncomplicated skin abscesses, trimethoprim-sulfamethoxazole does not decrease treatment failure at 7 days after incision and drainage.
• In adults with uncomplicated skin abscesses, trimethoprim-sulfamethoxazole might decrease the formation of new lesions at 30 days after incision and drainage
Suscribirse a:
Entradas (Atom)